Article Text

Abstract
Background Recent reports of the utilisation of pyrvinium pamoate (PP), an FDA-approved anti-helminth, have shown that it inhibits pancreatic ductal adenocarcinoma (PDAC) cell growth and proliferation in-vitro and in-vivo in preclinical models. Here, we report about an ongoing phase I open-label, single-arm, dose escalation clinical trial to determine the safety and tolerability of PP in PDAC surgical candidates.
Methods and analysis In a 3+3 dose design, PP is initiated 3 days prior to surgery. The first three patients will be treated with the initial dose of PP at 5 mg/kg orally for 3 days prior to surgery. Dose doubling will be continued to a reach a maximum of 20 mg/kg orally for 3 days, if the previous two dosages (5 mg/kg and 10 mg/kg) were tolerated. Dose-limiting toxicity grade≥3 is used as the primary endpoint. The pharmacokinetic and pharmacodynamic (PK/PD) profile of PP and bioavailability in humans will be used as the secondary objective. Each participant will be monitored weekly for a total of 30 days from the final dose of PP for any side effects. The purpose of this clinical trial is to examine whether PP is safe and tolerable in patients with pancreatic cancer, as well as assess the drug’s PK/PD profile in plasma and fatty tissue. Potential implications include the utilisation of PP in a synergistic manner with chemotherapeutics for the treatment of pancreatic cancer.
Ethics and dissemination This study was approved by the Thomas Jefferson Institutional Review Board. The protocol number for this study is 20F.041 (Version 3.1 as of 27 October 2021). The data collected and analysed from this study will be used to present at local and national conferences, as well as, written into peer-reviewed manuscript publications.
Trial registration number ClinicalTrials.gov: NCT05055323.
- Clinical Trial
- Hepatobiliary tumours
- Molecular aspects
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This study is the first of its kind to clinically assess pyrvinium pamoate as an anti-cancer therapy.
The study design of 3+3 single-dose escalation with two planned dose escalations allows assessment of multiple drug dosing protocols.
The protocol follows patients for 30 days after surgery and therefore not suitable to monitor oncological outcomes and instead assesses secondary molecular endpoints to measure tumor response.
This is a surgical ‘window of opportunity’ trial, which allows assessment of novel drug therapies without added confounders from concomitant standard chemotherapy, and with very early time span to assess molecular effects on the tumors.
The study is a single-institution study which limits generalisability from the population, facility and treatment protocol standpoints.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive and chemoresistant tumour that is associated with poor prognosis and 5-year survival rates of less than 10%.1 2 The pancreatic tumour microenvironment (TME) is hypovascularised, hypoxic and poor in nutrients. This leads to an exploitable vulnerability in the form of energy restriction. We have recently shown mitochondrial inhibition to have great potential in in-vitro inhibition of PDAC.3 Currently, no mitochondrial inhibitors have been approved by the FDA for cancer treatment. However, several drugs that have been shown to have mitochondrial targeting are approved for clinical use in other indications. Pyrvinium pamoate (PP) is a mitochondrial inhibitor, which has been approved by the FDA for use as an anti-helminthic agent. It has been shown in preclinical studies to act as an anti-cancer drug in breast, bladder and colorectal cancers,4–12 and was shown recently to have significant impact on pancreatic tumour growth in in-vivo mouse studies.3
Previous studies performed in humans that examined the safety and maximum tolerated dose (MTD) of PP showed that PP has no bioavailability at 5 mg/kg, after a single oral dose,13 while another study showed different patient cohorts were able to tolerate PP at 15–25 mg/kg, oral, for 5 days, and 35 mg/kg, oral, for 3 days, with no major side effected noted, with 3/34 patients complaining of abdominal pain.14 However, it is important to note that a dose of 35 mg/kg has not been studied beyond 3 days. Currently, there is only one well-known bioavailability study; however, the data are variable and over 20 years old.13 An update to the literature via newer studies are needed to better understand the bioavailability and pharmacokinetic and pharmacodynamic (PK/PD) of PP at both the maximum and minimum tolerated dosages.
In preclinical models, our lab has shown that PP is able to inhibit tumour growth both in vitro and in vivo.3 In these preclinical models, PP dosed at 1 mg/kg three times a week was found to lead to significant tumour growth inhibition and increased overall survival.3 In vitro we have shown that PP has <100 nM IC50 in many pancreatic cancer cell lines, patient-derived xenograft lines and patient-derived organoid lines.3 Additionally, in vivo, we have shown that PP is able to attenuate tumour growth both via intraperitoneal administration at 1 mg/kg, every other day, and at 5 mg/kg, 20 mg/kg and 35 mg/kg, oral, daily. In our in vivo experiments, we have found PP to be safe and with oral dosing there is a dose-dependent decrease in tumour growth, with best effects noted at 35 mg/kg, oral, daily dosing.3 In mice models, we have performed PK and biodistribution studies and have found that though PP has no detectable levels in the plasma after a single dose, it does have detectable and therapeutic levels in the fat and pancreatic tissue of mice dosed and can be detected 2 hours after a single dose of PP. Additionally, PP was not detected in the muscle tissue after this single dose, showing preferential distribution of PP in the fat.
In previous studies, PP has been shown to be tolerated at doses of 20 mg/kg and 35 mg/kg for 3 and 5 days, respectively, when used in the treatment of pinworms.13 15–21 The primary aim of this clinical trial is to assess the safety and tolerability of PP after three consecutive doses in patients with pancreatic cancer as this will help identify the MTD. Preclinical data have previously shown the potential of PP to synergise with chemotherapeutic agents for the treatment of pancreatic cancer.22 The PK/PD profile of PP and bioavailability in humans serves as the secondary aim.
Currently, patients with PDAC currently have few effective options for treatment. In light of previous preclinical success in the usage of PP as an anti-cancer agent for breast, bladder colorectal and pancreatic cancer, we believe that PP has the potential to be clinically beneficial in PDAC if found to be safe and tolerable in this patient population.
Methods/design
This is a single-centre window-of-opportunity phase I study. Reporting is based on the ‘Standard Protocol Items: Recommendations for Interventional Trials’ statement (https://www.spirit-statement.org/). The trial registration dataset is provided in the online supplemental materials.
Supplemental material
Study objectives
The primary objective of safety and tolerability will be measured via the observed dose-limiting toxicity (DLT) reported by the patient through daily phone conversations with a research coordinator. The secondary objective of the PK/PD profile will be measured through collecting blood and plasma levels 24 hours after 3 consecutive days of taking PP (day of surgery) and post-final PP dose on the day after surgery. Biopsy of pancreatic cancer tissue, normal pancreatic tissue and omentum will be taken and analysed using liquid chromatography followed by mass spectrometry (LC/MS). The pancreatic cancer tissue specimen will also be analysed for biological targets known to change with PP therapy, such as Ki67 and phospho-Retinoblastoma (pRb), electron transport chain (ETC) proteins, and mitochondrial RNA abundance.3
Study design and hypothesis
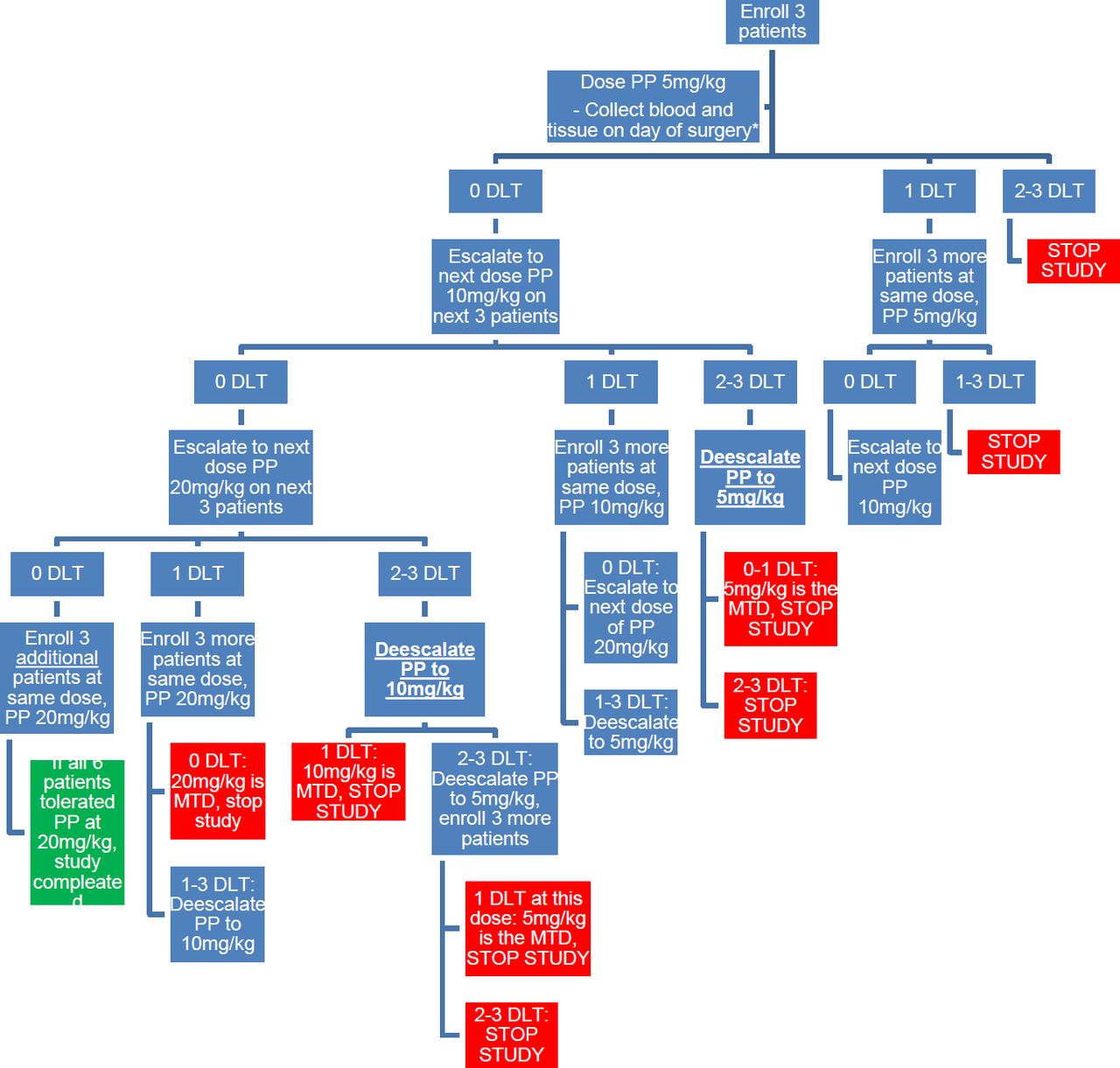
This study is a single institution, single-arm, open-label, phase I oncology trial with a single MTD dose-escalation study for PP. The study follows a 3+3 single-dose escalation design with two-dose escalations (figure 1). The number of participants ranges from 3 to 18, with an expected number of 12 participants if there are no DLTs. PP will be taken, for a total of 3 consecutive days of therapy, with the final day being the day before surgery. Patients will be monitored for a total of 30 days from the last dose to look for any side effects related to therapy.

Schematic of study design in a 3+3, dose escalation methodology. PP, pyrvinium pamoate; DLT, dose-limiting toxicity.
The initial dose of PP will be given at 5 mg/kg orally for 3 days for the first three patients. The next set of patients will be dosed at 10 mg/kg orally for 3 days if the initial dosage is tolerated. The final group of patients will be dosed at 20 mg/kg orally for 3 days if prior dosages are tolerated. Follow-up will occur every week to determine if any DLTs have occurred and will continue until 4 weeks after the first dose of PP. The time from initial patient accrual to a final analysis of data is expected to span from 12 to 18 months. DLT grade ≥3 is used as the primary endpoint. Dependent on DLT occurrence, dose de-escalation is also possible (figure 1) to further validate MTD. Therefore, since the sample size is based on the 3+3 design, the minimum number of participants is 3 (if the first dose is not safe), while the maximum is 18 participants. The expected sample size if there are no toxicities at the lower doses is 12 participants.
Participants
Inclusion criteria: individuals must meet all of the following inclusion criteria in order to be eligible to participate in the study:
Male or female, age>18, with PDAC who are deemed to be surgical candidates by the Thomas Jefferson University surgery department.
Patients will be assessed by the pancreatic surgeons in the pancreatic surgery clinic, and if they are found to have resectable disease, they can be considered for this study.
Patients must not be on neoadjuvant therapy or have received their last neoadjuvant treatment greater than or equal to within 3 weeks of starting PP therapy.
Provide signed and dated informed consent form.
Willing to comply with all study procedures and be available for the duration of the study.
Patients must have an estimated life expectancy of >3 months and Eastern Cooperative Oncology Group performance status of 0–1.
All patients, regardless of age or gender, must agree to observe proper contraceptive methods to avoid becoming pregnant or causing pregnancy for the duration of the study (30 days after the last dose of the drug).
Males will practice safe sex methods (eg, condoms).
Women of childbearing potential will practice safe sex methods (eg, condoms, birth control); if a female in the study is of childbearing age, they will have to take a urine hCG (pregnancy) test prior to enrolling in the study.
Exclusion criteria: an individual who meets any of the following criteria will be excluded from participation in this study:
Patients with ongoing anti-cancer therapies or those who will have received an anti-cancer therapeutic <3 weeks prior to the first dose of PP.
Any condition that precludes pancreatic surgical resection at the time of the study.
Pregnancy or currently breast feeding.
Known allergic reactions to components of the study product(s): PP/pyrvinium embonate (Molevac).
Patients with chronic bowel conditions (such as Irritable Bowel Syndrome [IBS]).
Kidney function impairment (serum creatinine >1.5× upper limit of norm (ULN) or creatinine clearance ≤60 mL/min/1.73 m2 for patients with creatinine levels >1.5× ULN).
Patients with liver function impairment: alkaline phosphatase, ALT and AST above threefolds the normal limit (see normal ranges below); total bilirubin level>3 mg/dL; albumin levels <3 g/dL.
If any participant is unable to continue with the study, or withdraws, then another participant that meets the inclusion and exclusion criteria will be chosen and reassigned. If the patient misses any of the three doses due to anything other than toxicity caused by the study drug, they will be excluded for the portion of the study relevant to toxicity for determining the safety of the medication, and will be replaced by another subject. If the patient is unable to complete surgery or only receives partial surgical intervention (ie, open close due to metastatic disease) due to a non-drug-related cause, the safety and tolerability data from these patients will still be included for the determination of drug safety.
Study intervention
The study product being used in this clinical trial is available as an over-the-counter suspension of PP (50 mg/5 mL), which is sold commercially under the trade name ‘Molevac’ (Infectopharm GmbH, Germany). PP can be stored at room temperature and is stable at this temperature for 2 years. The drug will be kept at the Thomas Jefferson University Investigational Drug Service (IDS) for the duration of the trial, where weight-based calculations and distributions will be calculated. The IDS will prepare for each patient personal, premeasured vials of PP suspension that contain the calculated daily dose.
Adverse effects
PP product inserts and safety datasheets state that most of the side effects fall under the ‘very rare’ category (<1 in every 10 000 cases); however, some gastrointestinal side effects fall under the ‘common’ category (1–10 users in every 100 cases). The gastrointestinal side effects include nausea, vomiting, bloating and diarrhoea or constipation. Due to the drug’s formulation, there is a chance that the stool can change colours or have a reddish colour after taking pyrvinium. Patient populations that should not take PP include infants less than 3 months, pregnant patients or patients that are breast feeding, and patients with impaired kidney or liver function.
Study compliance
Each patient will keep a drug diary for the duration of their participation in the trial. Compliance will be determined by the drug diary and a verbal confirmation from the patient on the day of surgery. Patients will be excluded from the study and replaced with another subject if they are found to miss any of the PP doses for reasons outside of toxicity.
Study schedule
A brief schematic of the study schedule is seen in figures 2 and 3. The screening visit occurs during pretreatment (visit 1, day −28 to −1). In this visit, informed consent is obtained and documented. A review of the patient’s medical records, medication history and presurgical workup is obtained; these are reviewed to determine eligibility based on inclusion/exclusion criteria. If consented, the patient is given PP (Molevac) to take at home during study days 0–3. At the patient’s home (visit 2, days 0–2), the patient is instructed to take PP (Molevac) at their assigned dosage orally for 3 days consecutively. The final day of treatment is the day prior to surgery. A drug diary will be kept by the patient, where they will record compliance with drug administration.

Layout of study design by visit. PP, pyrvinium pamoate; AE, adverse event.

{kind=link}
{kind=link}
{kind=link}
Schedule of procedures and interventions. DLT, dose-limiting toxicity.
On the day of surgery (day 4), the patient will be met by an investigator to discuss and record adverse events/DLTs. The investigator will record standard presurgical physical examinations, ensure compliance with PP dosage and blood will be collected to establish a baseline preoperative PP level. On the day post surgery (day 5), blood will be drawn to measure PP plasma levels. The final visit (visit 4, day 30±1) includes recording the adverse events/DLTs reported by the participant or observed by an investigator. Each patient will be seen in the clinic on day 30 after their last treatment, where a final assessment of any PP-related DLTs will take place.
Any patient that withdraws or terminates participation will be monitored for any DLTs via a questionnaire, clinic visit or phone call for a total of 30 days from the last dose of PP.
Statistical analysis plan
In order to calculate the MTD from the 3+3 single dose-escalation study design, the DLTs per drug dosage will be examined. The safety and tolerability data will be used for statistical analysis. Included in the analysis for determining drug safety are patients who receive partial surgical intervention or patients who are unable to complete surgery unrelated to the drug. Descriptive analysis will be performed on the PK/PD, bioavailability, fatty tissue accumulation of PP, plasma concentrations and all other outcomes.
Data collection and management
Study staff will maintain appropriate medical and research records for this study, in compliance with International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH E6), and regulatory and institutional requirements for the protection of confidentiality of participant information. Study staff will permit authorised representatives of Sidney Kimmel Cancer Center (SKCC) and regulatory agencies to examine (and when required by applicable law, to copy) research records for the purposes of quality assurance reviews, audits and evaluation of the study safety, progress and data validity. During the study, a RedCap database will be created to record deidentified data details on each clinic visit, along with relevant laboratory data collected.
Interim analyses and stopping rules
No interim analysis is planned. Planned stopping points will be determined by the 3+3 MTD single dose-escalation study design and if two or more DLT out of three patients (or six patients in the highest tolerated dose category). In any particular cohort observed, the dose will be de-escalated, or at the lowest dose, the study will be stopped if there are two or more DLTs observed.
Study endpoints
Primary: the primary outcome of safety and tolerability will be measured by observing DLT, which is considered any grade 3 or higher adverse events due to the drug itself or delay of surgery. Using a 3+3 protocol design, if any DLTs are encountered, the dose will either decrease or remain the same until MTD is determined. See the study design flow chart for more details (figure 1).
Secondary: mouse studies have shown that with a single dose of PP, at both 20 mg/kg, oral, and 35 mg/kg, oral, there are no detectable levels of PP in the plasma, but 2 hours after oral dosing, there are high levels of this drug in the fat and pancreatic tissue of mice which is cleared rapidly.3 We believe due to the lipophilic structure of this compound, despite its decreased bioavailable, it will accumulate in the fatty tissues like the pancreas. Our secondary endpoint seeks to assess PP levels in the pancreas tissue (both normal and cancerous tissue) after three consecutive days of dosing. Additionally, we will assess known downstream molecular effects of the drug by assessing the expression of pRb, KI67, ETC proteins, and mitochondrial RNA expression levels. All the secondary endpoints will be tested on samples that would normally be taken from the patients as part of their surgical care.
Future use of stored specimens and other identifiable data
No specimens or other identifiable data will be stored after the study has been completed.
Study oversight, auditing and monitoring: in addition to the PI’s responsibility for oversight, study oversight will be under the direction of the SKCC’s Data and Safety Monitoring Committee (DSMC). The SKCC DSMC operates in compliance with a Data and Safety Monitoring Plan (DSMP) that is approved by the National Cancer Institute (NCI), and independent of the PI/sponsor and possible competing interests. Clinical site monitoring and auditing are conducted to ensure that the rights of human participants are protected, that the study is implemented in accordance with the protocol and/or other operating procedures, and that the quality and integrity of study data and data collection methods are maintained. Monitoring and auditing for this study will be performed in accordance with the SKCC’s DSMP developed by the SKCC DSMC. The DSMP specifies the frequency of monitoring, monitoring procedures, the level of clinical site monitoring activities (eg, the percentage of participant data to be reviewed) and the distribution of monitoring reports. Some monitoring activities may be performed remotely, while others will take place at the study site(s). Appropriate staff will conduct monitoring activities and provide reports of the findings and associated action items in accordance with the details described in the SKCC DSMP.
Quality control and quality assurance
All participants will follow the protocol as stated above, and QA/QC along with compliance with drug therapy will be monitored by the clinical research coordination staff. During the study, staff will monitor compliance with drug therapy. All specimen track logs will contain a participant ID to prevent dissemination of participant personal information and all ID and participant information will be collected and secured with password-protected files only accessible by study personnel as well. No training methods are necessary for this study. If there is any deviation from the protocol, the study PI will be notified immediately. If there are any concerns with quality assurance/quality control (QA/QC) or any problems with data entry, the study PI will be notified immediately as well.
Trial status
This study is registered on ClinicalTrials.gov # NCT05055323. The Thomas Jefferson IRB protocol number for this study is 20F.041 (Version 3.1 as of October 27, 2021). Recruitment for this study began 29 October 2021, and it is expected to be completed on 1 May 2024.
Ethical standard
The investigator will ensure that this study is conducted in full conformity with the principles set forth in The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research, as drafted by the US National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (18 April 1979) and codified in 45 CFR Part 46 and/or the ICH E6. The protocol, informed consent form(s), recruitment materials and all participant materials will be submitted to the IRB for review and approval. Approval of both the protocol and the consent form must be obtained before any participant is enrolled. Any amendment to the protocol will require review and approval by the IRB before the changes are implemented in the study.
Informed consent process
Informed consent is a process that is initiated prior to the individual agreeing to participate in the study and continues throughout study participation. Extensive discussion of risks and possible benefits of study participation will be provided to participants and their families, if applicable. A consent form describing in detail the study procedures and risks will be given to the participant. Consent forms will be IRB approved, and the participant is required to read and review the document or have the document read to him or her. The investigator or designee will explain the research study to the participant and answer any questions that may arise. The participant will sign the informed consent document prior to any study-related assessments or procedures. Participants will be given the opportunity to discuss the study with their surrogates or think about it prior to agreeing to participate. They may withdraw consent at any time throughout the course of the study. A copy of the signed informed consent document will be given to participants for their records. The rights and welfare of the participants will be protected by emphasising to them that the quality of their clinical care will not be adversely affected if they decline to participate in this study. The consent process will be documented in the clinical or research record. In this study, any participant who meets the inclusion criteria stated above, and is 18 or older will be included in the study. This excludes children only, but any races or genders that meet inclusion criteria can be included in the study. Women who are pregnant or lactating will be excluded, as the study drug is not approved in these types of patients.
Participant confidentiality
Participant information will only be collected if consent to participate is obtained. This confidentiality is extended to cover the testing of biological samples and genetic tests in addition to any study information relating to participants.
The study protocol, documentation, data and all other information generated will be held in strict confidence. No information concerning the study data will be released to any unauthorised third party without the prior written approval of the sponsor.
Data handling and record keeping
The investigators are responsible for ensuring the accuracy, completeness, legibility and timeliness of the data reported. All source documents must be completed in a neat, legible manner to ensure accurate interpretation of data. The investigators will maintain adequate case histories of study participants, including accurate case report forms (CRFs) and source documentation.
Data collection and accurate documentation are the responsibility of the study staff under the supervision of the investigator. All source documents and laboratory reports must be reviewed by the study team and data entry staff, who will ensure that they are accurate and complete. Unanticipated problems and adverse events must be reviewed by the investigator or designee.
During this study, all data collected will be recorded in a secure password-protected database, RedCap, and only study personnel will have access to it. Any paper copies of data as a backup will remain in a locked, secure container.
The data to be collected include vital signs and physical exams of any participants during the study and follow-up time points. The laboratory data collected from these blood samples will be analysed by Alliance Pharma and will be secured in a password-protected file that only the company and study personnel have access to. All samples will have a deidentified ID before being sent to biochemical analysis to protect participant confidentiality. Safety data and any DLT encounters will be stored in RedCap along with vital signs and physical exams, which will all be deidentified and a password-protected secure database. All data will be collected and analysed at the end of the study, and a final study report will be generated to summarise the study findings. No interim report will be generated during this study. The data analysis for PK/PD parameters will be done in an unblinded fasion but deidentified to participant information, as the dose of drug administered is necessary to calculate relevant PK/PD parameters.
The data collected in this study, including any identifying information, will only be available to approved investigators. To maintain confidentiality and in accordance with Health Insurance Portability and Accountability Act (HIPAA) regulations, only deidentified data will be shared with outside investigators. Requests for data sharing will require the permission of the principal investigator.
Study records retention
Study records will be maintained for at least 3 years from the date that the grant federal financial report is submitted to the NIH. Study documents will be retained for a minimum of 2 years after the last approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or until at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product. These documents will be retained for a longer period, however, if required by local regulations. No records will be destroyed without the written consent of the sponsor, if applicable. It is the responsibility of the sponsor to inform the investigator when these documents no longer need to be retained.
Future use of stored specimens and other identifiable data
No specimens or other identifiable data will be stored after the study has been completed.
Patient and public involvement
Patients or the public were not directly involved in the design, or conduct, or reporting, or dissemination plans of our research. However, the plans for the study and notice of study activation were presented on the 15th and the 16th pancreatic cancer patient symposiums at Thomas Jefferson University, Philadelphia (November 2020, November 2021).
Ethics and dissemination
This study was approved by the Thomas Jefferson IRB committee (protocol number #20F.041, Version 3.1 as of October 27, 2021). This study will be conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice (ICH E6), the Code of Federal Regulations on the Protection of Human Subjects (45 CFR Part 46), and Thomas Jefferson University research policies.
The data collected and analysed from this study will be disseminated via presentations at local and national conferences, as well as peer-reviewed manuscript publications
Trial registration and data sharing statement
This trial was prospectively registered on ClinicalTrials.gov on 6 July 2021. The registration number is NCT05055323. Deidentified study data and results will be posted to the study’s ClinicalTrials.gov site.
Discussion
PDAC is an aggressive and lethal disease. Despite intensive research, we still mostly rely on DNA damaging drugs as a mainstay of oncologic therapy. However, pancreatic tumours frequently display resistance to these common chemotherapies and overall response rates of standard of care regimens such as FOLFIRINOX and Gemcitabine-nab Paclitaxel result in response rates of 32% and 23%, respectively.23 24
The pancreatic TME comprises unique features. It is hypovascular, desmoplastic, hypoxic and poor in nutrients.25 This distinctive extracellular milieu results in relative energy restriction and lends itself to be exploited as a possible metabolic vulnerability.26 27 As PDAC is characterised by metabolic reprogramming,28 29 interventions aimed to target key metabolic processes are being actively investigated as potential anti-cancer therapies for PDAC (NCT02514031, NCT00096707, NCT03504423, NCT00741403, NCT03699319).
PP is a safe, oral medication clinically used in treatment of pinworm infections.20 22 Over the past decade, numerous reports have indicated PP to have anti-cancer effects in vitro and in vivo, in a multitude of cancer cell lines.3 5 9 12 It has been shown to act through multiple mechanisms including modulation of WNT pathway signalling,11 12 inhibition of the androgen receptor,7 competitive inhibition of the RNA-binding protein HuR30 31 and as a mitochondrial inhibitor.3 6 The main limitation in translating these effects into the clinical setting has been the reportedly low bioavailability of PP. Though data are scarce, available reports, based on plasma levels measurements, suggest that PP is not systemically absorbed in humans at 5 mg/kg.13 We have found in a mouse model that while plasma levels may be low/undetectable, PP redistributed rapidly to fatty tissues and can be identified there after oral therapy.
Therefore, we have designed this current dose escalation study, starting with the commonly used PP dose of 5 mg/kg and going up to 20 mg/kg to assess safety and bioavailability of oral PP treatment. Due to the short course of treatment (3 days), we selected to assess molecular secondary endpoints rather than macroscopic parameters such as tumour size, which are not likely to be significantly affected over this short time period. The molecular targets chosen are KI67, pRB and ETC protein expression. Additionally, we will extract and measure mitochondrial-encoded RNA targets, which were shown to be significantly decreased on PP treatment.
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank the Jefferson Pancreatic Tumor Registry (JPTR), the Sidney Kimmel Cancer Center (SKCC) clinical research team, and our participants for their involvement in the trial.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
FMP and CWS contributed equally.
HL and AN contributed equally.
Contributors FMP, CWS, AN and HL wrote the first draft. BEL, CWS, JP, WBB, JRB, CY, AN and HL contributed to study design and methodology. BEL was involved with the statistical design and analysis. SC and TY contributed to research coordination, ongoing patient enrolment and outpatient research follow-ups. All authors contributed to editing the manuscript. All authors read and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.