Article Text

Abstract
Introduction Alport syndrome (AS) is one of the most common fatal hereditary renal diseases in human, with a high risk of progressing to end-stage renal disease without effective treatments. Mesenchymal stem cells (MSCs) have recently emerged as a promising therapeutic strategy for chronic kidney disease. However, the safety and therapeutic potential of MSC transfusion for patients with AS are still need to be confirmed. Therefore, we have designed a clinical trial to evaluate the hypothesis that intravenous infusion of human umbilical cord-derived MSC (hUC-MSC) is safe, feasible, and well-tolerated in children with AS.
Methods and analysis We report the protocol of the first prospective, open-label, single-arm clinical trial to evaluate the safety and preliminary efficacy of hUC-MSC transfusion in children with early-stage AS. Paediatric patients diagnosed with AS who have persistent albuminuria will be candidates for screening. Twelve eligible patients are planned to recruit and will receive hUC-MSC infusions under close safety monitoring, and complete the efficacy assessments at scheduled follow-up visits. The primary endpoints include the occurrence of adverse events to assess safety and the albuminuria level for efficacy evaluation. Secondary endpoint assessments are based on haematuria and glomerular filtration measurements. Each patient’s efficacy endpoints will be evaluated against their baseline levels. Additionally, the underlying mechanism of hUC-MSC therapy will be explored through transcriptomic and proteomic analysis of blood and urine samples.
Ethics and dissemination The protocol (V.1.0, date 17 January 2015) was approved by the institutional review board of the Affiliated Taihe Hospital of Hubei University of Medicine (ethical approval 03 March 2015). Written informed consent will be obtained from the patient and/or guardians before study specific process. In addition to publication in a peer-reviewed scientific journal, a lay summary of study will be available for participants and the public on the Chinese Organization for Rare Disorders website (http://www.cord.org.cn/).
Trial registration number ISRCTN62094626.
- Clinical Trial
- Paediatric nephrology
- Mesenchymal Stem Cells
Data availability statement
Data are available upon reasonable request. Data are available upon reasonable request to the principal investigator.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Human bio-samples will be collected for transcriptomic and proteomic analysis to provide deeper insights into the therapeutic mechanism of human umbilical cord-derived mesenchymal stem cells.
Several clinical sites will participate in the trial to address the anticipated difficulties in recruitment due to the rarity of Alport syndrome as a hereditary condition.
Conducting renal biopsies after treatment for pathological evaluation presents invasiveness and practical challenges.
Introduction
Alport syndrome (AS) is one of the most common human fatal hereditary renal diseases. Its prevalence is reported as 1 in 5000–50 000 live births.1 2 A deficiency of type IV collagen chains α3, α4 or α5 in the basement membranes of renal glomerulus, eyes and ears is caused by mutations in COL4A3, COL4A4 or COL4A5, respectively.2 Therefore, AS is characterised by haematuria, albuminuria and a progressive decline of renal function,3 as well as sensorineural hearing loss4 and ocular abnormalities.5 Patient outcomes depend on whether the disease is X-linked or autosomal.6 7 X-linked disease caused by COL4A5 mutations makes up 85% of cases.6 Moreover, male patients with COL4A5 deficiency are at high risk to develop end-stage renal disease (ESRD). Current treatments for patients with microalbuminuria focuses on symptom relief using angiotensin-converting enzyme inhibitors7 8 or angiotensin receptor blockers3 9 10 to reduce proteinuria and delay ESRD. However, long-term safety and efficacy in young children with early microalbuminuria require further study.11 Other novel therapeutic strategies focus on chronic inflammation12 13 and fibrosis14 induced by abnormalities of type IV collagen. The anti-inflammatory and anti-fibrosis therapies15 are still in research.16–18 Recently, mesenchymal stem cells (MSCs) have emerged as a promising therapeutic strategy for chronic kidney disease (CKD) attributing to their anti-inflammatory and anti-fibrosis properties. Preliminary benefits have been observed in preclinical and clinical studies of lupus nephritis,19 20 diabetic nephropathy21–23 and focal segmental glomerulosclerosis.24 For AS, the therapeutic potential of MSCs had been shown in previous studies using COL4A3 deficient mice.25 26 As patients with COL4A5 deficiency make up the majority of cases and face a high risk of developing ESRD, we have conducted a preclinical study using COL4A5 deficient mice to evaluate the therapeutic potential of human umbilical cord mesenchymal stem cell (hUC-MSC). Relief of albuminuria and longer life times were shown in the MSC-treatment group with significant alleviation of nephritis and fibrosis. Further clinical trials are still needed to confirm the safety and therapeutic potential of hUC-MSC transfusion for patients with AS. Therefore, we plan to conduct a single-arm, first-in-human trial to investigate the safety and preliminary efficacy of hUC-MSC in paediatric patients with early-stage AS. We expect that the findings of our study will provide the first clinical evidence about the safety and preliminary efficacy of hUC-MSC in children with AS.
Methods and analysis
Study objective, design and setting
The main objective of this study is to evaluate the hypothesis that intravenous infusion of hUC-MSC is safe and well-tolerance for patients with AS. The current study is designed as a single-arm, open-label clinical trial to evaluate the safety and potential benefits of hUC-MSC infusion in paediatric patients with AS. The study was planned in January 2015, with recruitment starting on 15 January 2023. We aim to recruit 12 participants by 31 December 2025 in this first-in-human trial to primarily confirm the safety profile. The prevalence of AS remains unknown in China. For this preliminary investigator-initiated study, the sample size was estimated based on the potential patient pool established in our study on the natural history of this inherited rare disease and expected dropout rates of 15%~20%. Within the limits of available research funding, we aim to enrol as many subjects as possible in light of potential clinical benefits and the favourable safety profile observed in our previous cerebral palsy studies.27 28 To address the hurdles of recruitment due to the limitation of patient population, multiple investigational sites are selected at the beginning of the study, and the recruitment phase could prolong if it is necessary on ethical approval. Additionally, basket trials are recommended for clinical studies of rare diseases with different subtypes but similar underlying biology, according to guidelines from the National Center for Drug Evaluation.29 Therefore, a basket trial design is employed in this pilot study to collect as much information as possible on safety and preliminary efficacy in more patients with AS without restriction on genetic subtypes. The study is planned to close on 31 December 2026. Patients are recruited from Affiliated Taihe Hospital of Hubei University of Medicine, Shenzhen Second People’s Hospital and Hainan Women and Children’s Medical Centre in China. Safety and potential clinical benefits will be evaluated by comparing outcomes after hUC-MSC therapy to baseline. Based on our preclinical research, the underlying mechanisms of hUC-MSC to relieve albuminuria are indicated to involve anti-inflammation and anti-fibrosis. Therefore, exploratory research is also designed to evaluate local inflammation and fibrosis in the kidney microenvironment based on biological samples (figure 1). Our protocol is organised according to the Standard Protocol Items: Recommendations for Interventional Trials checklist30 (online supplemental material).
Supplemental material

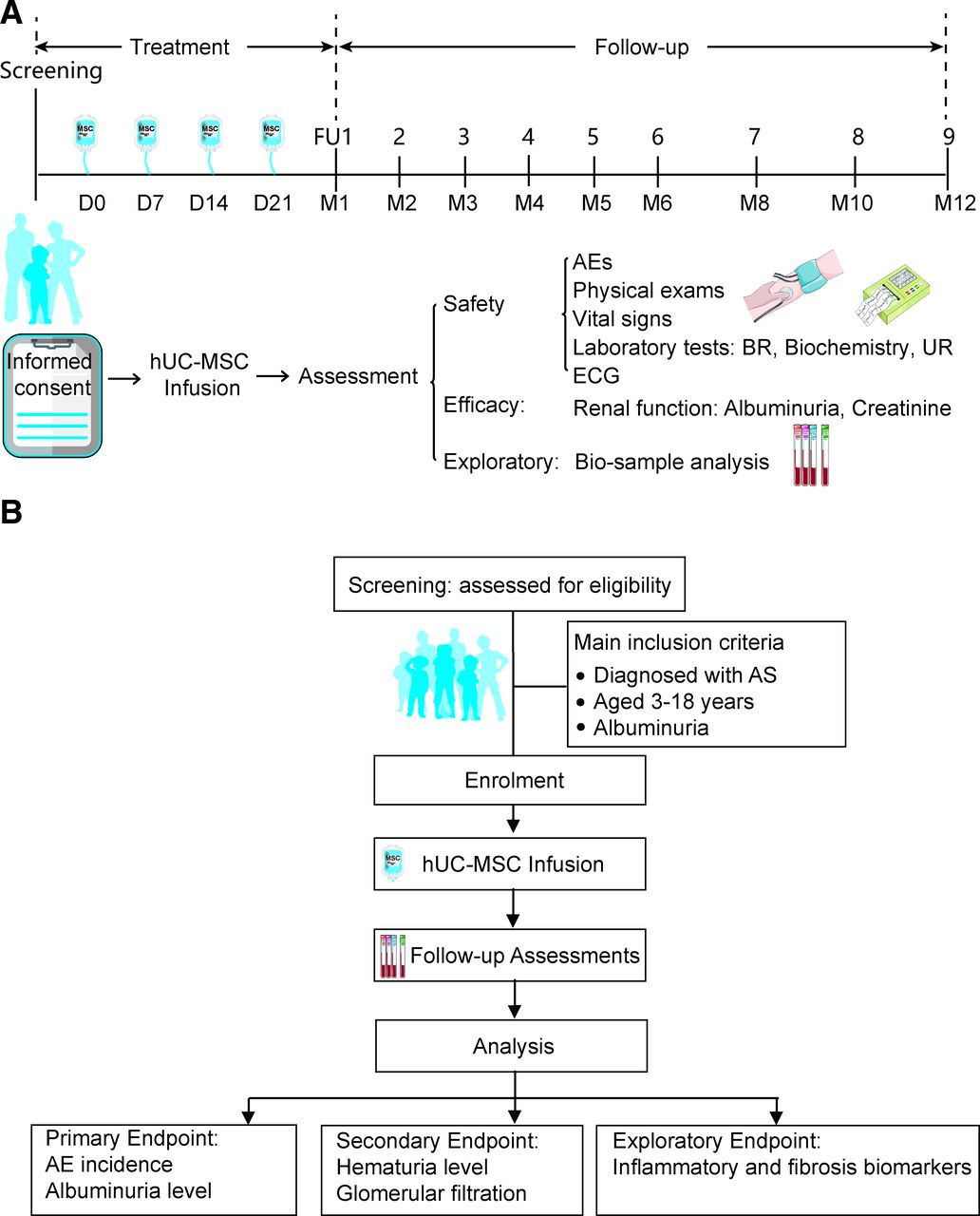{kind=link}
Study flow diagram. (A) The eligible patients will receive four doses of hUC-MSC infusion, and accept safety and efficacy assessments at scheduled visits. Blood and urine samples will be collected for exploratory research. (B) Occurrence of adverse events will be calculated as safety primary endpoint, while albuminuria will be evaluated and calibrated by creatinine for each patient against the baseline levels based on 24-hour urine samples. AEs, adverse events; AS, Alport syndrome; BR, blood routine test; ECG, electrocardiogram; FU, follow-up; hUC-MSC, human umbilical cord mesenchymal stem cell; UR, urine routine test.
Recruitment and eligibility criteria of subjects
Patients will be recruited from collaborative sites in South China. Written informed consent will be obtained from the patients and/or guardians before any study-specific procedures. The Doctor-Patient Community in AS plays an essential role in subject recruitment and compliance management to improve adherence to study procedures.
Patients of any gender who meet the following criteria will be included:
Diagnosed with AS according to the diagnosis criteria.3
Aged 3–18 years.
Albuminuria or combination with haematuria persisted without remission after routine treatments for 1 year.
CKD stage I-III, glomerular filtration rate >60 mL/min/1.73m2.
Written informed consent is obtained before study specific procedure.
Based on our previous preclinical research, MSC treatment alleviated albuminuria along with reduced inflammation and fibrosis. Therefore, rather than targeting specific genetics, the potential therapeutic mechanism of MSC may involve modulating inflammation and fibrosis, general pathological processes of CKD regardless of genotype variations in type IV collagen chains α. Thus, patients with AS with albuminuria could benefit most from this treatment. For this study, our target population is patients with AS with albuminuria without restriction of genotypes. While long-term outcomes are different between male and female patients with X-linked AS, they are similar for autosomal AS.6 31 Patient gender is not restricted in our study as we focus on short-term (1 year) safety and efficacy on albuminuria. No evidence from other MSC studies in CKD shows gender bias in safety or efficacy assessments. Long-term follow-up is planned for safety and clinical outcomes assessments in further extension study given the permission of the ethical committee.
Patients with one of the below conditions will be excluded:
Aged >18 years.
Creatinine clearance rate <30 mL/min, or CKD stage IV-V.
With one of the disease histories: immunological disease or autoimmune diseases; serious haematological or coagulation disorder; urogenital abnormalities; malignancy history; congenital heart disease or serious cardiovascular, liver or pulmonary dysfunction.
Uncontrolled endocrine diseases (eg, diabetes, hyperthyroidism).
Serious allergy history or known allergy to more than two kinds of foods or medications.
Active systemic infection.
Any other concerns that hampered compliance or safety as judged by the investigator.
Study treatments
Patients received hUC-MSC infusions at the Affiliated Taihe Hospital of Hubei University of Medicine, the leading site. Other treatments to manage symptoms are permitted and documented in medical charts, including antibiotic therapy for bacterial infection, and antipyretic medicine for fever.
hUC-MSC preparation
Allogeneic hUC-MSCs were purchased from Shenzhen Beike Biotechnology Company. As described in our previous studies,27 28 healthy postpartum women were screened according to the donor requirements of the American Association of Blood Banks32 for umbilical cords donation. Informed consent was obtained from eligible donors prior to sample collection. Cells were then isolated using sterile tissue-explant culture techniques. Cultured in serum-free Dulbecco’s modified Eagle’s medium supplemented with cytokines, the harvested cells were collected at passage 4 for flow cytometric analysis (FACSCalibur, BD Biosciences, San Jose, California, USA) to confirm expression of MSC-specific surface markers CD73, CD90 and CD105 over 95% and less than 2% expression of CD14, CD34, CD45 and CD79a/CD19. Osteogenesis, chondrogenesis and adipogenesis differentiation potentials were also evaluated. Quality tests as stipulated in International Society for Cellular Therapy standards33 34 were performed on each cell batch before release, including but not limited to viability, endotoxin, bacterial, fungal, mycoplasma and viral assays. The multipotency of MSCs were also tested for ossification, chondrification and adipogenesis, and received a quality certification of the National Institutes for Food and Drug Control (quality report serial number SH201401061), the top certification authority for stem cell quality in China.
hUC-MSC infusion
In our preclinical study, hUC-MSCs were injected via the tail vein at a fixed dosage of 5×106 cells for Col4a5 mutant AS mice at 6–8 weeks postnatal according to previous study using chorionic stem cells,25 UC-MSCs24 and bone marrow-derived stem cells (BMSCs)26 35 in Col4a3 mutant mice. While physiological, pharmacokinetic and toxicology data are recommended for interspecies scaling rather than simple BSA conversion to optimise drug development,36 the pharmacokinetic processes of stem cells in the human body, especially in children, remain unclear. Currently, no ideal method exists to translate stem cell doses used in animals to humans. Therefore, the hUC-MSC dosage was mainly based on previous clinical studies of other CKD. For adult patients with severe refractory systemic lupus erythematosus, their proteinuria was relieved after a single intravenous infusion of 1×106 cells/kg bone marrow-derived or UC-derived MSCs.37 For safety concerns due to differences in pharmacokinetics and pharmacodynamics between children and adults, 0.5×106 cells/kg was considered for dosage estimation in our study. Thus, a dosage range of 11–25×106 cells was calculated for children aged ≥7 years38 with body weights of 22–51 kg based on standard body mass index.39 Moreover, the dosage was calculated as 20×106 cells for patients aged around 12 years, a major group in our patient pool. No nephrotoxicity was observed in patients with rheumatoid arthritis receiving hUC-MSC infusion at a dose of 20×106 cells.40 Additionally, multiple hUC-MSC infusions at a fixed dose as 50×106 cells were safe in paediatric patients aged 3–12 years in our previous studies.27 28 Therefore, multiple infusions with a fixed dose of 20×106 cells were adopted in our study. This dosage is expected to provide a valuable reference for further dose-escalation studies.
Based on the safety data from previous studies,27 28 pretreatments with immunosuppressants will not be used. Patients will receive hUC-MSC infusion in a room equipped with resuscitation facilities at the Cell Therapy Centre. Before peripheral vein infusion, a quick quality check will be performed at the Cell Therapy Centre to ensure a viability rate exceeding 90% and an appropriate cell count of 20×106. The hUC-MSCs will be injected via peripheral vein after being dispersed in 50 mL of normal saline. The infusion will last 20–30 min. To monitor safety, the patient’s vital signs and consciousness status will be closely checked during the infusion, with special attention to fever and peripheral blood counts. Patients will receive four doses of hUC-MSC, with a 7-day interval between doses, provided there is good tolerance of the initial dose.
Study procedures and data collection plan
Study specific examinations will be conducted for screening after obtaining informed consent (online supplemental table S1). Baseline demographics, medical history and concomitant medications will be collected. Genetic test results and renal biopsy reports will be reviewed during screening to confirm the diagnosis. Clinical features will be evaluated based on physical examinations, vital signs, electric response audiometry and optical examinations. ECG and laboratory tests, including haematology, biochemistry, urinalysis, urine creatinine and albuminuria, will also be conducted for baseline assessment. Specifically, a 24-hour urine sample will be collected to evaluate renal function. Subject eligibility will be determined based on baseline assessments.
Supplemental material
Assessments for safety
A favourable safety profile was demonstrated in preclinical toxicology tests for the hUC-MSC product, including assessments of acute toxicity, long-term toxicity, haemolysis and tumorigenicity. Our previous studies have validated the safety of clinical application in children with cerebral palsy,27 28 although transient fever may occur based on other MSC transfusion studies.41–43 As the first trial to evaluate tolerance of hUC-MSC infusion in children with early-stage AS, safety evaluation through adverse event (AE) monitoring remains critical. The occurrence of AEs will be calculated as a primary endpoint. AEs are defined according to International Conference on Harmonisation-Good Clinical Practice (ICH-GCP) guidelines and identified via patient self-reports and examinations at scheduled visits following initial hUC-MSC infusion (online supplemental table S1). AE severity is assessed using the Common Terminology Criteria for Adverse Events (V.5.0) from the US Department of Health and Human Services. AEs and serious AEs (SAEs) will be documented and reported as required by ICH-GCP guidelines and local health authorities. To manage safety risks of hUC-MSC transfusion, the study will be conducted at several certified tertiary hospitals under the supervision of local health authorities. Additionally, patients will receive hUC-MSC infusion in a resuscitation-equipped room at the Cell Therapy Centre, with close safety monitoring during treatment.
Assessments for preliminary clinical efficacy
The urinary albumin-to-creatinine ratio detected from 24-hour urine samples will be assessed as the primary endpoint for efficacy assessment, based on our preclinical study in mice modelling AS, which demonstrated ameliorated albuminuria. Albuminuria level will be calibrated by creatinine value. Specifically, the urinary albumin and creatinine values will be obtained from the same 24-hour urine sample, and the ratio will be calculated by dividing the urinary albumin value by the creatinine value. Secondary endpoints will include assessments of haematuria and glomerular filtration rate. Urine red blood cell counts will be measured to evaluate haematuria, especially in patients with haematuria at baseline. Creatinine clearance (Ccr) will also be calculated to assess glomerular filtration function. Blood and urine samples will be collected at baseline, weekly during treatment and monthly in the follow-up period for testing (online supplemental table S1). Treatment-associated changes in albuminuria, haematuria and Ccr will be evaluated to assess efficacy.
Exploratory assessments
Renal biopsies for histopathological examination are difficult to perform after treatment due to their invasive nature. As an alternative approach, blood and urine samples will be collected for biomarker analysis using transcriptomics and proteomics to explore the underlying mechanisms of hUC-MSC therapy. Blood and urine samples will be collected from eligible patients before the first hUC-MSC dose, and at 7, 14 and 21 days after initial infusion. During follow-up visits, samples will also be collected 1 and 2 months after the last dose. Transcriptomics and proteomics analysis will be performed to examine changes in biomarkers before and after hUC-MSC therapy. Samples will be stored and destroyed in accordance with local health authority requirements
Withdrawal and termination from the study
Patients may withdraw from the study primarily due to AEs, lack of efficacy, major protocol violation, lost to follow-up or withdrawal of consent. Additionally, investigators may terminate patients based on a risk-benefit assessment in scenarios such as: poor tolerance of investigational therapy, SAE or other serious complications, rapid disease progression or deterioration of comorbidities. Patients who withdraw from or are terminated form the study should undergo a comprehensive assessment at their final visit. The causes of withdrawal or termination should be recorded in medical records.
Statistical analysis and data management
The analysis strategy is based on intention-to-treat (ITT) principle. For withdrawal or premature terminated cases due to AEs or poor efficacy, data will be included in the AE analysis or efficacy analysis, respectively, in line with the ITT principle. No imputation will be used for missing data. Statistical analysis will be conducted using SPSS software (V.20.0, International Business Machines Corporation, Armonk, New York, USA). Statistical data will be presented as the SD and range or median, quartiles and range for continuous variables and as frequencies and percentages for categorical variables. AE occurrence will be calculated to assess safety. Each patient’s efficacy endpoints will be evaluated against their baseline levels. Statistical significance is considered if p<0.05. Stratified analysis would be performed based on the CKDstage of the patients.
An interim analysis of safety and primary outcomes will be triggered once the first subject completes his 1-year follow-up visit or when four subjects have at least 1 month of safety data following hUC-MSC treatment, whichever comes first. The principal investigators and an independent data monitoring committee (DMC) will access the interim analysis results. Previous studies have shown an expected attributable SAE rate of less than 5% for hUC-MSC infusion in children with cerebral palsy,27 28 and favourable safety for MSC infusion in renal dysfunction.44 45 The study will be suspended if over 10% (1 of 12 subjects) experience an SAE related to hUC-MSC treatments. Study modification or termination may be considered for safety concerns, with the principal investigator making the final decision based on the DMC’s analysis and opinion. The DMC includes biostatisticians from Hubei University of Medicine and members of the ethics committee, who are independent of the study sites and free from conflicts.
To improve the data quality, a data management team was established for this multicentre trial, including study coordinator at local site for data collection and biostatisticians at the leading site for data analysis. Double data entry will be performed at the leading site, where two researchers cross-check the data for quality control and communicate with site staff to resolve any data queries. Additionally, the study was registered in the national clinical trial registry as required by local health authority. Progress of the current study is tracked through annual reports submitted to the registry. Auditing by local health authority, independent of study sites and investigational staff, is also accepted.
Patient and public involvement
Patients and their families are not involved in the study design or other scientific aspects of project management. However, they are kept informed of annual study progress through activities led by non-profit AS community groups. The funder was not involved in the study design, data management, data interpretation, report writing or submission.
Ethics and dissemination
Ethical approval was obtained from the institutional review board (IRB) of the leading site, Affiliated Taihe Hospital of Hubei University of Medicine (ethical approval 03 March 2015). Other sites accepted the leading site’s ethical approval and collaborated in patient recruitment and follow-up. The IRB at all sites tracked study progress through periodic safety reviews, annual reports and reports of any major protocol deviations. The study is conducted according to the protocol and could be modified based on IRB-approved protocol changes. Written informed consent is obtained from patients and/or guardians by authorised investigators before any study specific procedures, including biological specimen collection. The risk and potential benefits are listed in the informed consent forms and are discussed with the patients and their guardians at screening visit, including the accessibility of ancillary care and insurance for participants in clinical trials. De-identified data are extracted from the medical databases and medical charts for analysis, and no individual information will be disclosed during or after the trial. The study has finished registration on the WHO international clinical trials registered organisation platform, ISRCTN registry, and the study specific information could be available in the register system (https://doi.org/10.1186/ISRCTN62094626). In addition to publication in a peer-reviewed scientific journal, a lay summary of study will be available for participants and the public on the Chinese Organization for Rare Disorders website (http://www.cord.org.cn/).
Data availability statement
Data are available upon reasonable request. Data are available upon reasonable request to the principal investigator.
Ethics statements
Patient consent for publication
Acknowledgments
We acknowledge the staff at multiple-centre investigational sites for their intellectual advices to improve the study process.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors LH and CZ contributed to study design and protocol draft. CZ took responsibility for investigational product management. JZ contributed to biostatistical planning. YZ contributed to bio-sample management. JZ, YZ, JG, JW and CZ contributed to resource and investigation. LH, JZ, JW and JG contributed to funding acquisition. LH drafted the manuscript and took responsibility for organisation of regulatory affairs in the study. CZ contributed to the review and editing of the manuscript, supervising the work and correspondence. All authors revised the manuscript for important intellectual content and gave final approval for publication. Professional writer is not employed for protocol drafting and study result publications.
Funding This study was in part supported by the Guangdong Basic and Applied Basic Research Foundation (Grant No.2023A1515220023), Shenzhen Second People's Hospital Clinical Research Fund of Shenzhen High-level Hospital Construction Project (Grant No.2023yjlcyj012), Shenzhen Key Medical Discipline Construction Fund (SZXK060), Hainan Province Clinical Medical Center (QWYH202175), and Science and Technology Research Project of Hubei Province (No. 2013BCB002).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.