Article Text

Abstract
Background In adult patients with high myopia (HM), progressive axial elongation poses a significant risk for the development of subsequent ocular complications that may lead to visual impairment. Effective strategies to reduce or prevent further axial elongation in highly myopic adult patients have not been available so far. Recent studies suggested that medically lowering intraocular pressure (IOP) may reduce axial elongation.
Objective This clinical randomised controlled trial (RCT) aims to evaluate the efficacy of medical IOP reduction in adult patients with progressive HM (PHM).
Trial design Single-centre, open-label, prospective RCT.
Methods This RCT will recruit 152 participants with PHM at the Zhongshan Ophthalmic Center (ZOC). Randomised in a ratio of 1:1, participants will receive IOP-lowering eyedrops (intervention group) or will be followed without treatment (control group) for 12 months. Follow-up visits will be conducted at 1, 6 and 12 months after baseline. Only one eye per eligible participant will be included for analysis. The primary outcome is the change in axial length (AL) within the study period of 12 months. Secondary outcomes include the incidence and progression of visual field (VF) defects, changes in optic disc morphology and incidence and progression of myopic maculopathy. Difference in AL changes between the two groups will be analysed using linear regression analysis. For the secondary outcomes, a multifactor Poisson regression within a generalised linear model will be used to estimate the relative risk of progression in VF defects and myopic maculopathy, and the rate of thinning in retinal nerve fibre layer and ganglion cell-inner plexiform will be assessed through Kaplan-Meier curves and log-rank tests.
Ethics and dissemination Full ethics approval for this trial has been obtained from the Ethics Committee of ZOC, Sun Yat-sen University, China (ID: 2023KYPJ110). Results of this trial will be disseminated through peer-reviewed journals and conference presentations.
Trial registration number NCT05850936.
- randomized controlled trial
- ophthalmology
- glaucoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
Prospective, interventional, randomised controlled trial to evaluate the effect of intraocular pressure-lowering medications for controlling axial elongation in adult patients.
Designing and performing this randomised controlled trial based on an on-going large-scale highly myopic cohort.
Study does not include a placebo group and blinding is not applicated.
Participants are recruited from a single centre.
Introduction
High myopia (HM) is an important global public health issue,1 2 with prevalence estimates of approximately 163 million individuals (2.7% of the world population) affected in 2000, and of approximately 938 million (9.8% of the world population) people estimated to be affected in 2050.3–5 High myopia-related complications, such as optic neuropathy, myopic maculopathy and retinal detachment, can lead to irreversible visual impairment.6–11
Recent clinical studies have revealed that highly myopic eyes in adult patients can undergo further axial elongation with a rate of 0.1 mm/year.12 Axial elongation in highly myopic eyes is a major risk for progression of myopic macular degeneration and potentially of high myopia-associated optic neuropathy and subsequent vision impairment.13 14 Effective strategies to reduce or stop further axial elongation in highly myopic eyes are warranted.
Recent experimental studies have suggested that medical reduction of intraocular pressure (IOP) could be protective against axial elongation in guinea pigs.15 16 In a clinical observational study, the application of IOP-lowering medication, but not the IOP value itself, was associated with a reduced ongoing axial elongation in highly myopic patients.17 As a corollary, recent Mendelian research has established a bidirectional association at the genetic level between myopia and primary open-angle glaucoma mediated through IOP.18 In a recent retrospective clinical study medically IOP-lowering reduced axial elongation in highly myopic eyes (own unpublished data).
Building on these findings, we hypothesise that medically IOP-lowering may slow axial elongation by potentially three pathways related to the sclera and choroid.19 We therefore aim to conduct a randomised controlled trial (RCT) to assess the efficacy of medical IOP-lowering in managing axial elongation in patients with progressive HM (PHM). Additionally, this study should generate data on the effects of IOP-lowering treatment on the incidence and changes in the visual field (VF), changes in the optic nerve head morphology and myopic maculopathy. The outcomes of the study may establish a basis for treatment recommendations for preventing axial elongation of highly myopic eyes.
Methods and analysis
Study design
The PHM study is an open-label, single-centre RCT. The study will be conducted at the Zhongshan Ophthalmic Center (ZOC), Sun Yat-sen University, a tertiary specialised hospital in Guangzhou, China. All examinations and interventions will be carried out in the Clinical Research Center at ZOC. This study does not permit blinding and is therefore designed as an open-label trial. Figure 1 summarises the design of the PHM study.

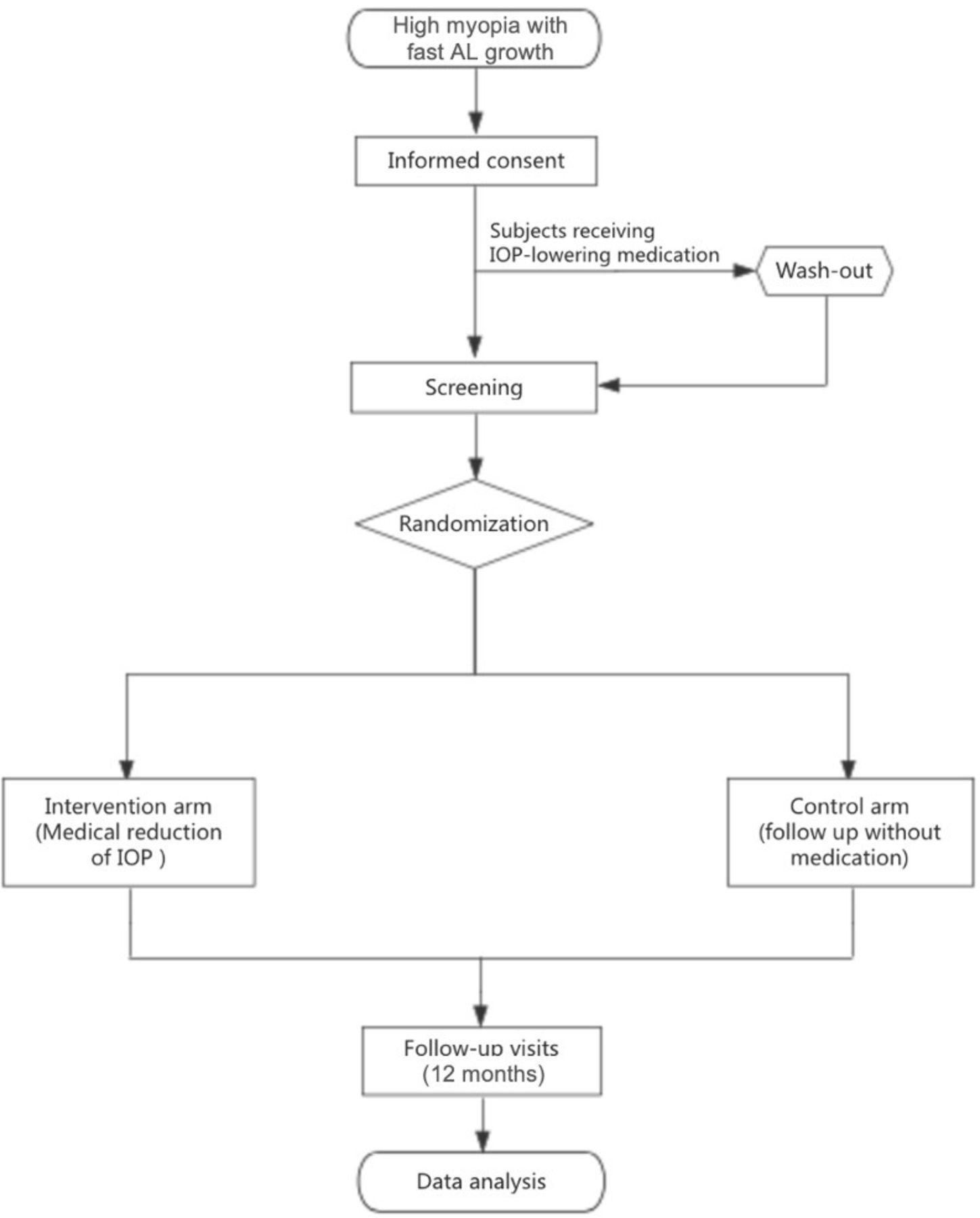{kind=link}
Diagram of the progressive high myopia study design. AL, axial length; IOP, intraocular pressure.
Objective
The primary aim of this trial is to evaluate the effectiveness of medically IOP-lowering therapy in managing axial elongation in patients with PHM over a 12-month observation period. Additionally, the trial will assess alterations in VF, optic disc morphology, thickness of the retinal nerve fibre layer (RNFL) and retinal ganglion cell-inner plexiform layer (GC-IPL) and the occurrence or advancement of myopic maculopathy.
Recruitment
Inclusion criteria
Age ≥18 years and ≤65 years.
Diagnosed with HM:20 21 spherical equivalent ≤−6.00 dioptres or axial length (AL)≥26.5 mm.
Diagnosed with PHM: axial elongation ≥0.05 mm in the past 6 months or ≥0.1 mm in the past 12 months.
IOP: ≥10 mm Hg and ≤21 mm Hg on at least two visits using Goldmann applanation tonometry with correction for the dependence of the IOP-reading on corneal thickness.22
Best corrected visual acuity (BCVA)≥6/12, ability to undergo AL measurement, fundus photography, optical coherence tomography (OCT) and complete VF examination.
Exclusion criteria
Patients who have been using IOP-lowering medications within the last year.
Allergy to any kind of IOP-lowering eyedrops.
Presence of serious fundus pathologies like proliferative diabetic retinopathy, retinal detachment, central retinal artery occlusion, etc.
Presence of chronic, recurrent or severe ocular inflammatory lesions such as chronic or recurrent uveitis.
Significant corneal or iris lesions, severe cataract affecting fundus examination or patients with only one eye.
Intraocular surgery or laser treatment within the last year, such as cataract surgery.
With a history of previous refractive surgery or prior treatment for myopia-related conditions (eg, orthokeratology lens wear, low-intensity red light therapy or low-concentration atropine treatment).
Presence of other serious systemic diseases (eg, hypertension, heart disease, diabetes, rheumatic immune system disease) that hinder long-term follow-up and eye treatment.
Pregnancy, lactation or plans to have children during the follow-up period.
In this study, only one eye per eligible participant will be included. If both eyes meet the inclusion criteria, the eye with a higher rate of axial elongation, a worse mean perimetric deviation value and a worse BCVA will be selected.
Randomisation and blinding
In this trial, randomisation will be employed to mitigate distribution bias. After confirming all inclusion and exclusion criteria and obtaining signed written informed consent forms (see online supplemental material), qualified individuals within each block, with a block size of 4, will be assigned in an even (1:1) manner to either the intervention group or the control group. The random sequence will be generated using an electronic data collection (EDC) system to ensure unbiased allocation.
Supplemental material
In this trial, the participants and physicians will not be blinded to the intervention assignment. The technicians conducting the examinations and interpreting the images will be unaware of the participants’ group assignments during the screening and follow-up stages. The researchers analysing the data will also be unaware of the information regarding randomisation.
Interventions
Intervention group
Participants assigned to the intervention group will receive medical IOP-lowering therapy for a duration of 12 months or until they reach the endpoint. Only the study eye will receive medication in the enrolled participants. The preferred medication for reducing IOP is Xalacom eye drop (Pfizer, New York, New Y, USA), a fixed latanoprost and timolol combination.
The treatment protocol will involve the instillation of a single drop of prostaglandin ophthalmic solution in the study eye once daily in the evening for medications such as Xalacom. To ensure the standardisation of medication usage among participants, subjects will be provided with medication logbooks, which will be collected and recorded by the investigators during the study visits.
Control group
Participants assigned to the control group will be followed-up for 12 months or until they reach the endpoint without medical IOP-lowering therapy.
Outcome measures
Primary outcome
The primary outcome is the change of AL at 12 months from baseline measured by IOLMaster.
Secondary outcomes
Incidence and progression of VF defects at 12 months from baseline based on Humphrey 24–2 standard VF. Under the premise of reliable VF examination, compared with the baseline, two consecutive perimetric examinations reveal the incidence of VF defects or significant perimetric progression in at least three points at a significance level of p<0.05. Furthermore, two subsequent diagnostic VF examinations conducted within 1 month also confirm the progression at the locations. The time of progression is defined as the time of the initial diagnostic VF examination.23 24
Changes in optic disc morphology (including thinning of RNFL and GC-IPL) at 12 months from baseline based on fundus photography and OCT.25–27
Incidence and progression of myopic maculopathy at 12 months from baseline based on fundus photography and OCT. The determination of the incidence and progression of myopic maculopathy is based on the META-PM classification system.28 29
Study assessments
Visual acuity
Visual acuity assessment will be conducted prior to any procedures that may potentially impact vision, such as pupil dilation or VF examination. The measurement of visual acuity will be performed using an ETDRS (Early Treatment of Diabetic Retinopathy Study) LogMAR chart (Precision Vision, Villa Park, Illinois, USA) under standard illumination conditions at 4 m.30 For BCVA, a trial frame will be positioned and adjusted on the participant’s face based on auto-refractometric readings and subsequent subjective refinement.
Refractometry
Following pupil dilation using 0.25% compound tropicamide (Zhuobian; Sinqi, China), three measurements will be taken for each eye using an auto refractometer (KR800, TOPCON, Tokyo, Japan). The average values for spherical refractive error, cylindrical refractive error and astigmatic axis will be recorded for further analysis and documentation.
Slit-lamp biomicroscopy
The evaluation of the anterior segment with the pupil undilated will be performed using a slit lamp (BQ-900, Haag Streit, Koeniz, Switzerland). After medical pupillary dilation, a slit lamp-based grading of lens opacities is conducted and using a 90D indirect ophthalmoscopic lens (Ocular 90D Slit Lamp Lenses, Ocular, Washington, DC, USA), the optic disc, macula and peripheral retina will be examined.
Tonometry
IOP measurements will be performed by Goldmann applanation tonometry (AT900, Haag Streit, Koeniz, Switzerland). Prior to enrolment, all participants will undergo three baseline IOP readings during specific time intervals: 09:00 to 10:00, 13:00 to 14:00 and 16:00 to 17:00. During follow-up visits, tonometry will be conducted between 09:00 and 11:00. Results from three consecutive measurements will be documented during each visit, and the mean value of these measurements will be used for assessment purposes.
AL measurement
AL measurement will be performed using the IOLMaster (IOLmaster 700, Carl Zeiss Meditec, Jena, Germany). Results from five consecutive measurements will be documented during each visit, and the mean value of these measurements will be used for further analysis.
Central corneal thickness measurement
Central corneal thickness measurement will be performed with IOLMaster (IOLmaster 700, Carl Zeiss Meditec, Jena, Germany). Results from five consecutive measurements will be documented during each visit, and the mean value of these measurements will be taken for further analysis.
Perimetry
The perimetric examination will be performed applying the Humphrey Field Analyzer Mark 3 (Carl Zeiss Meditec, Dublin, California, USA) and the Swedish Interactive Threshold Algorithm Standard 24–2 programme. A reliable VF report is defined as having false-positive and false-negative errors below 15%, as well as fixation losses below 20%.
Fundus photography
Using fundus cameras (KOWA, Nonmyd, WX3D, Nagoya, Japan; TRC-NW400, TOPCON, Tokyo, Japan), two fundus images centred on the optic disc will be taken for each eye under both standardised stereoscopic and non-standardised conditions. Additionally, a single image focused on the macula will be obtained after pupil dilation.
OCT examination
All participants will undergo a series of standardised swept-source OCT examinations using the DRI-OCT Triton model (TOPCON, Tokyo, Japan), focusing on the optic disc and macula. To ensure the reliability and accuracy of the results, a minimum image quality score of 60 will be set. In addition to the swept-source OCT, a spectral domain OCT examination will be conducted to obtain measurements of the peripapillary RNFL and the macular GC-IPL.
Pregnancy test
A urine pregnancy test will be performed for women of reproductive age during their initial visit.
Anthropometry and blood pressure
Participants’ height and weight measurements will be measured using a free-standing height rod and a calibrated scale (RGZ120, Jiangsu Wujin Weighing Apparatus Factory, Jiangsu, China). During the baseline visit, blood pressure readings will be obtained from the participant’s left arm while they are seated and have rested for a minimum of 5 min with the Omron M7 Blood Pressure Monitor (Matsusaka, Mie, Japan).
Visit schedule
Table 1 summarises the visit schedule for the enrolment, interventions and assessments of this trial.
Visit schedule
Sample size
The sample size calculation for this study is determined based on the primary outcome and the study hypothesis, taking into account relevant findings from previous studies. The objective of this study is to evaluate the potential of IOP-lowering therapy to reduce axial elongation during a study period of 1-year growth in eyes with PHM. It is hypothesised that the intervention group receiving IOP-lowering therapy will exhibit a 70% reduction in axial elongation compared with the control group. The control group is estimated to experience a 0.1 mm axial elongation over 12 months, while the intervention group is expected to have a mean axial elongation of 0.03 mm. The common SD is estimated to be 0.14 mm. To achieve a statistical power of 80% with a two-sided significance level of 0.05, a total of 64 individuals per group is required. Accounting for an estimated 15% loss to follow-up at the 12 months mark, the final sample size is determined to be 76 individuals per group, resulting in a total of 152 participants. The sample size calculation was performed using PASS V.16.0 software (NCSS, LLC, Kaysville, Utah, USA).
Statistical analysis
Statistical analysis will be conducted using Stata V.16.0 software (StataCorp, College Station, Texas, USA). A two-sided p value of <0.05 will be considered statistically significant, and a 95% CI will be used for parameter estimation.
For the intention-to-treat (ITT) analysis, missing data will be addressed using the multiple imputation method. No simulation will be performed for missing data in the safety evaluation. Dropout rates in the two groups will be compared using χ2 tests or Fisher’s exact tests. Descriptive statistics will be reported as mean and SD for normally distributed continuous data, and as median and IQR for non-normally distributed continuous data. Frequency and percentage will be provided for categorical data. Baseline data, including demographic and clinical characteristics, will be analysed using independent samples t-tests and Wilcoxon rank-sum tests for continuous data, and χ2 or Fisher’s exact tests for categorical variables.
The analysis of primary and secondary outcomes will follow the ITT principle, including all participants. For the primary outcome, the difference in axial elongation between the two groups will be analysed using linear regression analysis. As for the secondary outcomes, a multifactor Poisson regression within a generalised linear model will be used to estimate the relative risk of progression in perimetric defects and myopic maculopathy. Additionally, the rate of thinning of the RNFL and the GC-IPL will be assessed through log-rank tests.
Safety analysis will be performed on participants belonging to the intervention group, comparing adverse event occurrence between the two groups using the χ2 test or Fisher’s exact test.
The missing data will be handled using the multiple imputations method. With the multiple imputation approach, 20 replicas of the data set will be generated, where missing values are imputed through chained equations. The final results will be obtained by averaging these 20 data sets using Rubin’s rules.
Data monitoring
The Data Monitoring Committee (DMC) will closely monitor the data throughout the trial using the EDC system to ensure the reliability and integrity of the collected data. The DMC members are independent individuals who are not affiliated with the researchers or sponsors, ensuring an impartial assessment. There is no conflict of interest between the researchers and sponsors, further guaranteeing the transparency and objectivity of the data monitoring process.
Interim analysis: The study does not include provisions for conducting an interim analysis.
Data management
The collected data will be meticulously recorded and entered into the EDC system. The EDC system is securely hosted on a password-protected network server, ensuring digital protection. Only the principal investigators and authorised study team members will have access to the research data. To ensure confidentiality and integrity, all source documents will be stored in locked file cabinets with restricted access. Prior to data collection, all researchers will undergo comprehensive training. The raw data will be monitored by an independent Data and Safety Monitoring Committee. In the event of queries or uncertainties in the case report form, the data administrator will generate a data queue request (DRQ) and communicate the query to the researcher through the clinical monitoring system. The researcher is expected to provide a prompt response to the data administrator. If necessary, data modifications, confirmations and entries will be made, and a new DRQ will be issued.
Drug packaging, management, dispensing and storage
Drug packaging
The investigational drugs will be packaged in their own containers with dedicated research labels. Each medication package will consist of a single box, and each box will contain one unit of the drug. The box packaging will be made of paper and labelled with the phrase ‘For Clinical Research Use Only’, along with a drug label indicating a unique drug identification number. The drug identification number is composed of the first two letters of the drug name followed by a four-digit Arabic numeral.
Drug management
The investigator will directly purchase the investigational drugs through the ZOC Research Procurement System. The drugs will be received and stored by a dedicated medication administrator in the Clinical Research Center. The investigational drugs will be managed by the designated medication administrator, who will be responsible for:
Storing the drugs according to the storage conditions specified in the drug instructions.
Recording all drug dispensing and retrieval activities.
Dispensing the drugs only to the participants as specified in the research protocol.
Maintaining a comprehensive record of drug inventory throughout the study and providing inventory logs.
Maintaining a detailed catalogue of the drugs, including information on received materials, dispensing dates and records of drugs provided to participants.
Ensuring that the drug dispensing records match the usage and unused drugs and providing explanations for any discrepancies. Relevant dispensing and return forms must be signed by the medication administrator.
Drug dispensing
After randomisation, the medication administrator will dispense the corresponding investigational drugs to the participants based on the randomised assignment and document the drug dispensing.
Drug storage
Unopened medications should be stored according to the instructions provided in the drug package insert. For drugs that require refrigeration between 2°C and 8°C, the medication administrator needs to monitor the temperature and humidity daily and document the storage conditions. The research drugs should not be provided to anyone other than the participants in the study. Access to the research drugs is limited to personnel authorised by the principal investigator to dispense them. After opening, medications should be stored according to the instructions provided in the drug package insert and must be used within 4 weeks of opening the drug package.
Safety assessments
The safety assessments included in this study are as follows:
Medication-related safety assessment: The ocular hypotensive medications used in this study are known to have rare occurrences of local and systemic adverse reactions, such as ocular surface irritation, eyelid pigmentation, eyelash growth and drug allergies. These assessments will be conducted and recorded by the investigators during the study visits using slit lamp examination.
High myopia-related safety assessment: This study focuses on individuals with high myopia, and during the natural course of high myopia, retinal pathologies can occur. The investigators will assess and record the complications associated with high myopia based on the examination results during the study visits.
Report and management of adverse events
An adverse event refers to any negative medical occurrence experienced by a participant in the study, regardless of its relation to the treatment. Utmost attention will be given to identifying potential adverse events or unfavourable findings. The primary concern is the safety of the participant, and appropriate medical intervention will be provided in case of an adverse event. All adverse events, whether reported voluntarily by the participant or discovered through questioning, physical examination, or other means by the study staff, will be promptly recorded on an online adverse event form The Safety Supervision Committee will review each form to determine the appropriate coding and reporting procedures.
Serious adverse events, regardless of their connection to the study drug, must be reported within 24 hours to the Institutional Review Board, the DMC and the Clinical Research Center. Additionally, a faxed report must be sent to the drug administration’s drug registration office. The original and fax confirmation forms for serious adverse events should be retained in the research centre along with the case report form.
Discussion
This trial is designed to evaluate the effect of IOP reduction on progressive high myopia. Due to the limited research on the use of IOP reduction medications in highly myopic eyes, the design of this study is based on a retrospective analysis of a large cohort of highly myopic individuals. It was found that the use of IOP reduction medications slowed down axial elongation in approximately 70% of highly myopic eyes. Therefore, we performed sample size calculations based on these findings.
Study progress
The recruitment period of this study started in June 2023 in ZOC. As of February 2024, we have included 73 participants.
Ethics statements
Patient consent for publication
Acknowledgments
We thank all the members of the Glaucoma Suspects with High Myopia (GSHM) Study Group for conducting this trial. And also, we thank all staff in the clinical research centre of Zhongshan Ophthalmic Center for their effort in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JJ and TL contributed equally.
Collaborators GSHM study group: Principal investigators: Yizhi Liu, Lin Lv, David Friedman and Tin Aung. Members: Wei Wang, Fei Li and Kai Gao. Steering committee: Neil M. Bressler, Ki Ho Park, Mingguang He, Kyoko Ohno-Matsui and Robert N Weinreb. Data monitoring committee: Ching-Yu Cheng, Paul Healey and Linda M Zangwill. Safety supervision committee: Xiang Chen and Guangxian Tang.
Contributors XZ, FL, JBJ, XG and SC participated in the study design. JJ, TL and FL wrote the primary protocol manuscript. XZ, FZ, DSCL and JBJ revised the manuscript. KK, PW and YS contributed to data collection. LJ and WZ helped with sample size calculation and were the statistical consultants. YL, JC and MC were clinical research coordinators of the project.
Funding This article was supported by the National Key R&D Program of China (2022YFC2502800); the High-level Hospital Construction Project, Zhongshan Ophthalmic Center, Sun Yat-sen University (303020104); the National Natural Science Foundation of China (82070955); the Science and Technology Program of Guangzhou, China (202201020362, 202102010209 2024A03J00515); Natural Science Foundation of Guangdong Province (2020A1515011282). The funding organisations had no role in the design or conduct of this article.
Competing interests JBJ: European patent EP 3,271,392, JP 2021-119187 and US 2021 0340237 A1: Agents for use in the therapeutic or prophylactic treatment of myopia or hyperopia; European patent application 23196899.1 ‘EGFR Antagonists for the treatment of diseases involving unwanted migration, proliferation, and metaplasia of retinal pigment epithelium (RPE) cells’.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note TL is a visiting doctor at Zhongshan Ophthalmic Center.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.