Article Text

Abstract
Introduction Patent ductus arteriosus (PDA) is the most common cardiovascular problem that develops in extremely preterm infants and is associated with poor clinical outcomes. Uncertainty exists on whether early pharmacotherapeutic treatment of a clinically symptomatic and echocardiography-confirmed haemodynamically significant PDA in extremely preterm infants improves outcomes. Given the wide variation in the approach to PDA treatment in this gestational age (GA) group, a randomised trial design is essential to address the question. Before embarking on a large RCT in this vulnerable population, it is important to establish the feasibility of such a trial.
Methods and analysis Design: a multi-centre, open-labelled, parallel-designed pilot randomised controlled trial.
Participants: preterm infants born <26 weeks of gestation with a PDA diagnosed within 72 hours after birth.
Intervention (selective early medical treatment (SMART) strategy): selective early pharmacological treatment of a moderate-severe PDA shunt (identified based on pre-defined clinical signs and routine screening echocardiography) within the first 72 postnatal hours with provision for repeat treatment if moderate-severe shunt persists.
Comparison (early conservative management strategy): no treatment of PDA in the first postnatal week.
Primary outcomes: (1) proportion of eligible infants recruited during the study period; (2) proportion of randomised infants treated outside of protocol-mandated therapy.
Sites and sample size: the study is being conducted in seven neonatal intensive care units across Canada and the USA with a target of 100 randomised infants.
Analysis: the primary feasibility outcomes will be expressed as proportions. A pre-planned Bayesian analysis will be conducted for secondary clinical outcomes such as mortality, severe intraventricular haemorrhage, procedural PDA closure and chronic lung disease to aid stakeholders including parent representatives decide on the appropriateness of enrolling this vulnerable population in a larger trial if the feasibility of recruitment in the pilot trial is established.
Ethics and dissemination The study has been approved by the IWK Research Ethics Board (#1027298) and six additional participating sites. On the completion of the study, results will be presented at national and international meetings, published in peer-reviewed journals and incorporated into existing systematic reviews.
Trial registration number NCT05011149 (WHO Trial Registration Data Set in Appendix A).
Protocol version Ver 7.2 (dated July 19, 2023).
- Randomized Controlled Trial
- Neonatal intensive & critical care
- NEONATOLOGY
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This randomised trial exclusively enrols infants born less than 26 weeks of gestational age.
Unlike previous patent ductus arteriosus (PDA) trials, the SMART-PDA trial combines clinical and echocardiographic criteria to grade the severity of the PDA shunt to decide on the treatment in the intervention group rather than deciding based on the PDA size and shunt directionality only.
Given this is a pilot trial to assess the feasibility of recruitment, information obtained from this trial should not be used for clinical decision-making.
The trial uses ibuprofen as first-line pharmacotherapy and therefore is unable to assess the feasibility of recruitment in centres using indomethacin or acetaminophen as the first-line therapy for PDA.
Background
Patent ductus arteriosus (PDA) is the most common cardiovascular problem that develops in infants born extremely preterm and is associated with poor neonatal outcomes such as death, necrotising enterocolitis (NEC) or chronic lung disease (CLD).1–6 Randomised controlled trials (RCTs) exploring pharmacotherapeutic treatments for PDA have demonstrated that non-steroidal anti-inflammatory agents (indomethacin and ibuprofen) and acetaminophen are effective in closing a PDA.7 8 However, existing RCTs have not been able to demonstrate a reduction in PDA-related adverse outcomes.7–9 Yet, an overwhelming majority of clinicians continue to treat PDAs early in the smallest preterm infants.10
Evidence from observational studies suggests that an approach of early echocardiography screening and treatment might be beneficial in extremely preterm infants. In a French national population-based cohort of 1513 preterm infants, screening echocardiography and PDA treatment before postnatal day 3 in extreme preterms was associated with lower rates of in-hospital mortality and pulmonary haemorrhage.11 Similar results have been demonstrated in a recent study from Iowa that showed a 23% absolute reduction in the primary outcome of death or severe BPD (p=0.002) in the cohort of 22–24 week GA infants with early haemodynamic screening.12 This approach of early screening and treatment, however, has not been evaluated through adequately powered trials in infants at the highest risk of adverse clinical outcomes, such as those born <26 weeks of GA.
On the contrary, a conservative approach in the first postnatal week would lead to less exposure to potentially harmful medications as well as less resource use (less echocardiographic assessment). A recent RCT on early ibuprofen therapy for PDA in extremely preterm infants born <28 weeks of GA (the Beneductus Trial) not only demonstrated the non-inferiority of an expectant management approach versus early pharmacotherapy but, in fact, the composite outcome of death/NEC/CLD was worse in the early treatment group (absolute risk difference −17.6%; 95% CI, −30.2 to −5.0).13 It is not clear whether this reflects the elimination of an important role for the PDA in some patients or unanticipated harmful effects on the developing lung in those patients whose PDA was unresponsive to treatment. Therefore, an early conservative approach might be a safer option to manage PDA, especially in micropreemies at <26 weeks of GA, who are at a high risk of PDA-attributable morbidity as well as medication-related adverse effects.
Given the strong pathophysiological rationale of adopting either approach, there is always a risk of significant protocol violations while attempting to address this question through a large RCT. To our knowledge, no RCT on PDA management has ever been conducted that exclusively enrols micropreemies born at <26 weeks of GA. Two recent pilot RCTs on early PDA treatment within a similar timeframe (within 48 hours by El Khuffash et al from Ireland and within 72 hours by de Waal et al from Australia) have shown wide variation in recruitment rates (88% vs 54%, respectively).14 15 Furthermore, another recent multi-centre RCT on early targeted treatment of the PDA conducted in France (TRIOCAPI trial) showed that open-labelled treatment occurred in 62.3% of infants at a median age of 4 days, which substantially dilutes the trial results and interpretation.16 Therefore, a pilot trial to assess the feasibility of recruitment and protocol adherence is required before designing a large multi-centre trial.
Research question and objectives
The overall purpose of this pilot RCT is to assess the feasibility of conducting a large RCT to explore the following research question: “In preterm infants born <26 weeks’ GA, does a strategy of selective early treatment of a moderate-severe PDA shunt (based on pre-defined clinical and echocardiographic criteria) in the first postnatal week lead to reduction in the composite outcome of death or severe CLD when compared with an early conservative management strategy?”
The study objectives are as follows:
The primary objectives are to assess (a) the proportion of eligible infants recruited in the trial and (b) the proportion of randomised infants with treatment outside of protocol-mandated therapy.
The secondary objectives are to (a) compare clinical outcomes between the planned comparison groups, (b) views of parents/guardians on enrollment in this RCT and (c) assess the feasibility of conducting a cost-effectiveness analysis for the main trial.
Primary hypothesis for the pilot RCT
Recruitment of preterm infants born at <26 weeks of GA in a trial of selective early medical treatment versus conservative management of the PDA is feasible with minimal protocol deviation.
Methods
Study design
This is a multi-centre, open-labelled, parallel-designed pilot RCT. At the time of publication of this protocol, seven centres (four in Canada and three in the USA) have actively recruited in the trial. The protocol has been designed in accordance with the Consolidated Standards of Reporting Trials (CONSORT) extension for randomised pilot and feasibility trials and the Standard Protocol Items: Recommendations for Interventional Trials guidance for reporting clinical trial protocols.17–19 Recruitment for the study commenced on 10 January 2022, with a tentative completion date of September 2024.
Patient and public involvement in study design
A parent partner from the Canadian Premature Babies’ Foundation, which is a parent-led, non-profit organisation providing education, support and advocacy for Canada’s premature babies and their families, was involved in the design of the study including the development of eligibility criteria and prioritisation of outcome measures.
Eligibility criteria
Preterm infants born less than 26 completed weeks (ie, up to and including 25 weeks and 6 days) of gestation are eligible for enrolment following written informed consent from the parents/guardians antenatally or after birth (figure 1). All enrolled infants undergo a screening echocardiography within the first 72 hours of age. Infants with an open PDA (of any shunt severity) documented on the initial screening echocardiography, who do not satisfy any of the exclusion criteria described below, are potentially eligible for randomisation. Infants receiving prophylactic cyclo-oxygenase inhibitor therapy (indomethacin, ibuprofen or acetaminophen) are also eligible for inclusion if the screening echocardiography is performed after completion of the course of prophylactic cyclo-oxygenase inhibitor drug but before 72 hours of age.

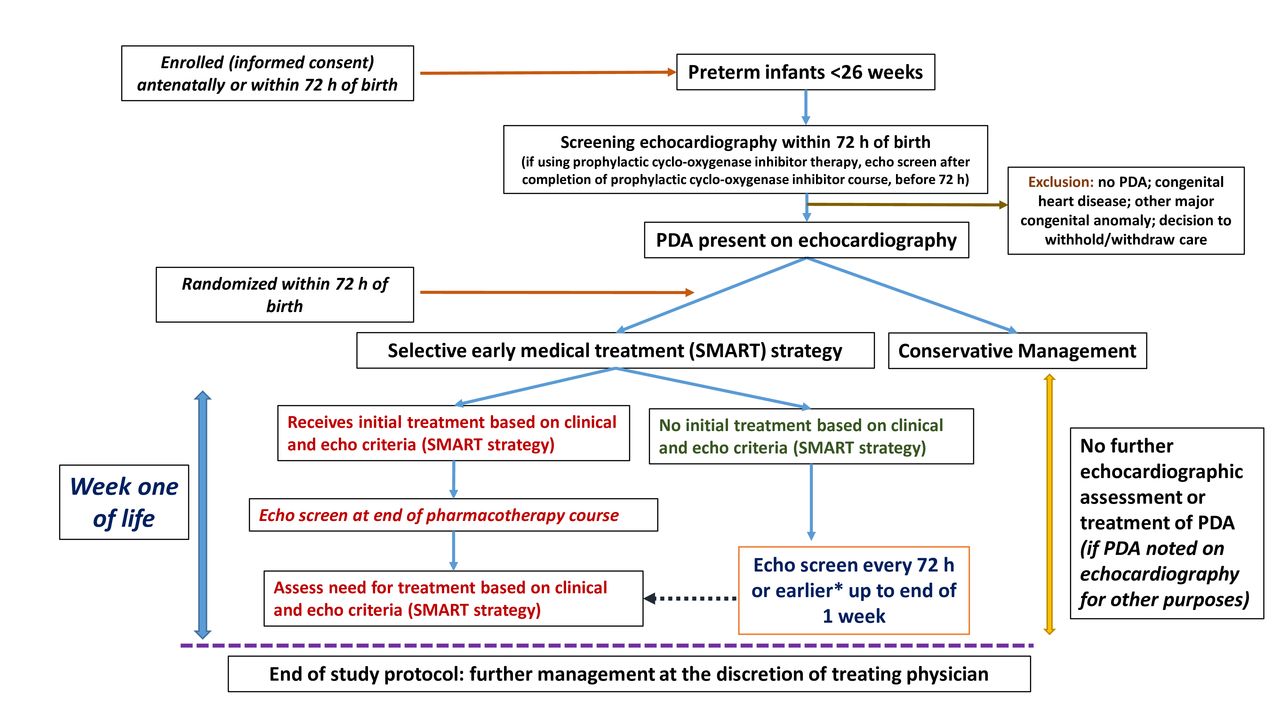{kind=link}
Study flow diagram. *If there is worsening clinical severity status. echo, echocardiography; PDA, patent ductus arteriosus.
Preterm infants born at <26 weeks of GA with an antenatally/postnatally diagnosed major congenital anomaly or congenital heart disease or infants where a decision has been made to withhold active management are not eligible for enrolment. Further, enrolled infants are not eligible for randomisation if (a) on initial screening echocardiography the PDA is closed or a major congenital heart disease is diagnosed (excluding patent foramen ovale, atrial septal defect or ventricular septal defect with a defect size of less than 2 mm) that precludes any PDA closure treatment or (b) a decision has been made to withdraw active management before randomisation.
Interventions
Experimental group (selective early medical treatment (SMART) strategy)
Infants randomised to the experimental group follow the SMART treatment protocol, which includes echocardiographic screening every 72 hours or earlier (if there is worsening clinical severity status) to categorise PDA disease severity by combining clinical and echocardiographic features (table 1). The PDA shunt severity is determined based on a combination of clinical and echocardiographic signs that have been adapted from validated classification systems and modified based on feedback from neonatologists practicing targeted neonatal echocardiography in Canada (table 1).20–23 All echocardiographic assessments are done by physicians trained in targeted neonatal echocardiography (TNE) or by paediatric cardiologists (based on echocardiography protocols of participating sites). Details of the standardised echo views and measurements are outlined in online supplemental appendix B.
Supplemental material
Clinical and echocardiographic measures of the haemodynamic significance of the patent ductus arteriosus (PDA)
At any evaluation if patients are found to have a ‘severe PDA’ on echocardiography, irrespective of clinical symptoms, or a ‘moderate PDA’ on echocardiography with at least moderate clinical illness, they receive pharmacotherapy aimed at PDA closure. All other combinations are managed conservatively (ie, without the use of NSAIDs or acetaminophen) (table 2). Additionally, if the PDA shunt direction is not predominantly left-to-right (ie, ≤66% of the cardiac cycle), no treatment is immediately provided as PDA closure may be detrimental in the presence of pulmonary hypertension. However, the infant is screened with echocardiography every 72 hours (or earlier if there is worsening clinical severity status) as the PDA may become haemodynamically significant as pulmonary pressures decline over time.
Approach to treatment in the intervention arm based on shunt volume and its clinical effects (SMART protocol)
Pharmacotherapy, when indicated (ie, for ‘severe PDA’ on echocardiography, irrespective of clinical symptoms, or a ‘moderate PDA’ on echocardiography with at least moderate clinical illness), is provided in the form of ibuprofen as a first-line agent at a standard dosing of 10 mg/kg followed by two doses of 5 mg/kg every 24 hours. The route of administration may be intravenous or enteral, as determined by the treating team. For treated infants, follow-up echocardiography is conducted at the end of the 3-day course, and a second course of treatment is initiated if they still fulfil study treatment criteria as mentioned above (table 2). If any treatment-eligible infant has a contraindication to ibuprofen as determined by the medical team, use of acetaminophen (15 mg/kg/dose every 6 hour for 3–7 days; intravenous or enteral) is permitted as an alternative agent.
Control arm (early conservative management strategy)
Infants randomised to this arm do not undergo any further echocardiographic assessment or any pharmacological treatment of the PDA regardless of their clinical signs. If the infant gets an echocardiographic assessment for a reason different than study-related PDA assessment (such as hypotension or oxygenation failure or as a part of a separate clinical study) and a PDA is incidentally noted that fits the treatment matrix (table 2), the infant is not initiated on pharmacotherapy. After 7 days of age, decision on PDA assessment and treatment is at the discretion of the treating physician (figure 1).
Safety parameters for considering rescue management in the control arm
The presence of severe life-threatening clinical signs in infants in the control group that may prompt an echocardiography evaluation and PDA treatment are as follows: (a) pulmonary haemorrhage defined as blood-stained respiratory secretions with an acute significant increase in respiratory requirements (mean airway pressure >12 cm H2O and/or fraction of inspired oxygen >60%);24 (b) persistent systemic hypotension defined as mean blood pressure (in mm Hg) below the GA (in completed weeks) for >30 min.24
Study procedure
Consent and enrolment
Parents/guardians of eligible infants are approached for informed consent antenatally or within 72 hours of birth (sample consent form in online supplemental appendix C) (figure 1).
Randomisation
The unit of randomisation is the infant. Eligible preterm infants are randomised in a 1:1 ratio using computer-generated random numbers in randomly permuted blocks of 4 or 6. The study coordinator for each site randomises the infant using a secure RedCap-based application and notifies the neonatal intensive care unit (NICU) team caring for the infant.
Blinding
This is a pragmatic open-labelled trial aimed at assessing the feasibility of recruitment. Therefore, the care providers are not blinded to the allocation and are allowed to use other NICU interventions in both arms as per institutional protocol. These co-interventions are recorded for comparison. However, to protect from detection bias, the outcome assessors for the secondary clinical outcomes are blinded.
Outcome measures
Primary feasibility outcomes
(1) The proportion of eligible infants recruited during the study period, (2) the proportion of treatment outside of protocol-mandated therapy among randomised infants and (3) the proportion of infants in the control group meeting pre-defined safety criteria.
Secondary feasibility outcomes
(1) Reasons for non-recruitment for eligible infants and non-adherence to protocol, (2) completeness of data collection for clinical outcomes, (3) qualitative views of parents on recruitment and (4) inter-observer and inter-centre reliability of echocardiography measurements.
Secondary clinical outcomes
(1) Mortality during hospital stay, (2) procedural PDA closure, (3) proportion of infants receiving any PDA pharmacotherapy, (4) proportion of infants receiving open-label rescue medical treatment, (5) CLD (defined as the need for supplemental oxygen or respiratory support at 36 weeks of postmenstrual age),25 (6) postnatal corticosteroid use for CLD, (7) pulmonary haemorrhage, (8) duration of invasive mechanical ventilation, (9) intraventricular haemorrhage (IVH; grades I to IV),26 (10) severe IVH (grades III and IV),26 (11) periventricular leukomalacia (any grade),27 (12) NEC (stage 2 or greater),28 (13) gastrointestinal bleeding within 7 days of the first dose of pharmacotherapy;, (14) spontaneous intestinal perforation, (15) severe retinopathy of prematurity (ROP) (stage 3 or greater),29 30 (16) blood culture-confirmed sepsis, (17) oliguria (defined as <1 mL/kg/hour) and (18) duration of hospitalisation (days).
Health economic outcomes
(1a) Timeliness of access to costing data, (1b) similarity of costing methods across sites2 and (2) evaluation of different cost-assessment approaches (ie, actual costs associated with care of infants in the trial, modelled costs based on historical data and resource use and modelled costs based on literature).
Sample size for pilot trial
Being a pilot study, the desired sample size was based on the sample required to reliably demonstrate feasibility. The criteria for success of the pilot study (feasibility measures) are defined in table 3.
Criteria for considering feasibility
Assuming a 70% recruitment of eligible infants and assuming a CI of 10% around this estimate to be acceptable,31 the required sample size to demonstrate feasibility (ie, the lower bound of CI >60%) will be at least 100 infants. Similarly, assuming an 85% protocol adherence of randomised infants, to demonstrate protocol adherence feasibility (ie, the lower bound of CI >75%), a minimum of 77 infants will be required. Therefore, success of the feasibility study can be reliably demonstrated with a total sample size of 100 randomised infants.
Based on the consensus of the trial steering committee, the pilot RCT will be deemed ‘definitely feasible’ if we are able to recruit more than 60% of eligible infants during the study period and protocol adherence is demonstrated in more than 75% of randomised infants (table 3). The pilot RCT will be deemed ‘may be feasible’ if we are able to recruit 40–60% of eligible infants during the study period and protocol adherence is demonstrated in 65–75% of randomised infants. If the pilot RCT is deemed ‘definitely feasible’ or ‘may be feasible’ and no further protocol changes are mandated by the trial steering committee, all 100 participants may be rolled-over into the full-scale SMART-PDA trial. If a decision is made by the steering committee to modify the trial protocol in any form for the full-scale SMART-PDA trial, then data from the SMART-PDA pilot trial will be analysed as a separate standalone trial.
Data management
Data collection plan
All trial data are documented on a pre-specified case report form (CRF) and entered on a trial specific database through RedCap with participants identified only by their unique trial number. The database has been developed and maintained by RedCap and the SMART-PDA Research Team. Access to the database is restricted and secure. Any missing or ambiguous data are queried, ideally within 2 weeks of the query being raised. For infants with missing data (eg, if an infant has been transferred to another hospital and data have not been obtained from the continuing care site), data are obtained from the receiving hospital where possible. A secure link to an online questionnaire (RedCap) is sent to parents before discharge of the infant from the NICU. For parents who do not have internet access or prefer to complete a paper copy, a paper version is provided.
Source data management
In order to allow for the accurate reconstruction of the trial and clinical management of the participant, source data will be accessible and maintained. Source data are kept as part of the woman’s and infant’s medical notes generated and maintained at the site. Each site records the location of source data at their site using a source data location log. Data that are not routinely collected elsewhere are entered directly onto a paper CRF workbook or RedCap; in such instances, the CRF workbook or RedCap acts as source data, which is clearly defined in the source data location log. For this trial, source data refer to, though is not limited to, the woman’s medical notes, infants’ medical notes, women’s and infants’ local electronic case records, infant’s echocardiography images and reports and parent questionnaires.
Data archiving
It is the responsibility of the principal investigator to ensure all essential trial documentation and source documents (eg, signed informed consent forms, investigator site files, participants’ hospital notes, copies of CRFs etc) are securely retained for at least 25 years after the end of trial. No documents will be destroyed without prior approval from the trial steering committee.
Statistical analysis plan
Eligibility, recruitment and retention through the study will be presented in a CONSORT flow diagram.17 The analysis will be by intention-to-treat with due emphasis on CI for between-arm comparisons. CIs will be interpreted within a descriptive compatibility framework and will not be used for inference or incorporated in a formal stopping rule. Descriptive statistics of demographic and clinical measures will be used to assess balance between the randomised arms at baseline, but no formal statistical comparisons will be made.
The primary feasibility outcomes will be expressed as proportions. For secondary clinical outcomes, a Bayesian analysis will be conducted to explore the posterior probability of benefit or harm, along with the corresponding 95% credible intervals (CrIs). Prior probabilities obtained from the most recent Cochrane review of early treatment versus expectant management of the PDA in preterm infants will be used for the Bayesian analysis.32 Our previous work demonstrated that parents view death, severe IVH, NEC and CLD as critical for their child’s well-being.33 Hence, we plan to develop a series of binomial models for each of these outcomes and then conduct a stochastic multicriteria acceptability analysis incorporating partial utilities estimated by swing weights for the said outcomes based on our previous work to determine the probability of net positive benefit with the SMART approach.34 The rationale for conducting the Bayesian analysis is to aid stakeholders including clinicians and parent partners decide on appropriateness of enrolling this vulnerable population in a larger trial with similar methodology if the feasibility of recruitment in the pilot trial is established. The posterior probabilities obtained from the Bayesian analysis is not intended to be used for clinical decision-making. A thematic analysis approach will be used to qualitatively analyse the views of parents on enrollment of their infants born at <26 weeks of GA in this trial.35
Given that estimation of effectiveness of the SMART protocol is not the primary objective of this trial, we do not plan to conduct any interim analysis or set any ‘statistical stopping rules’. Therefore, all analyses will be done at the end of the recruitment period. All secondary outcomes, in addition to the feasibility outcomes, will be reported for inclusion in meta-analyses updates. Table 4 summarises the analysis plan for all stated primary and secondary objectives.
Summary of the study objectives, outcomes and analysis plans
For the larger definitive trial, which will be informed by this pilot trial, assuming a baseline risk of 80% for death/CLD (2019 CNN data),25 to demonstrate superiority of the SMART approach over conservative management (10% absolute risk reduction of death/CLD), we will require a minimum of 588 infants (with a two-sided α of 0.05 and power of 80%).
Safety considerations
Data safety monitoring board (DSMB)
A DSMB has been set up comprising of two neonatologists and a biostatistician who are not study investigators and are free of any financial or intellectual conflicts of interest. The DSMB meets every 6 months (or earlier if deemed necessary) to ensure the overall safety of patients in the SMART-PDA pilot trial based on a review of the totality of evidence and the principle of the emergence of proof beyond a reasonable doubt that is likely to influence clinical practice, thereby minimising avoidable harm and maximising benefit.
Reporting of serious adverse events (SAEs)
For this study, an SAE is defined as one that results in death, is life-threatening or results in persistent or significant disability or incapacity. Potential SAEs include death, NEC (stage 2 or 3), gastrointestinal perforation and severe IVH (grades 3 and 4). Specific SAEs are recorded and reported to the institutional research ethics committee as soon as it occurs expeditiously, according to institutional regulatory reporting requirements for the duration of the study (36 weeks of corrected GA or discharge from the study site NICU, whichever is later). Reports on SAEs are also sent to the DSMB within 72 hours of diagnosis. In addition, all serious unexpected adverse drug reactions with respect to the use of ibuprofen or acetaminophen that has occurred inside or outside Canada are reported to Health Canada as follows:
If it is neither fatal nor life threatening, within 15 days after becoming aware of the information.
If it is fatal or life threatening, within 7 days after becoming aware of the information.
Trial management
Trial oversight is conducted by the Trial Steering Committee that is comprised of a team of experienced neonatal clinical trialists, early career investigators in neonatology, biostatisticians, a paediatric cardiologist and a parent partner representative. The members of the trial steering committee are listed in online supplemental appendix D. Safety of trial participants is monitored by an independent Data Safety Monitoring Board as mentioned above.
Trial monitoring
The trial is being carried out in accordance with the Declaration of Helsinki in its latest form and the International Conference on Harmonisation Good Clinical Practice (ICH-GCP) guidelines. The site investigator consents to data evaluation being performed by the monitoring team (comprising the PI and his delegates) to ensure satisfactory data collection and adherence to the study protocol. A summary of the protocol amendments to date and the corresponding explanations are outlined in online supplemental appendix E. The tasks of the site investigator include maintenance of the source data as comprehensively as possible; this includes information concerning medical history, accompanying diseases, inclusion in the trial, data about visits, results of imaging and investigations, dispensing of medication and adverse events. The monitoring team is also permitted to perform data evaluation and draw comparisons with the relevant medical files in accordance with the standard operating procedures and ICH-GCP guidelines at predetermined intervals to ensure adherence to the study protocol and continuous registration of data. All original medical reports required as sources for the information given in the CRF may be inspected. The substitute decision makers of the study participants will have given their consent to such inspection by signing the consent form. The monitoring team members are obliged to treat all information as confidential and to preserve the basic claims of the study participants in respect of integrity and protection of their privacy.
Anticipated challenges and solutions
We have thought of and planned for challenges likely to be faced in our study design and execution: ( 1) Risk of performance bias due to lack of blinding: we acknowledge that co-interventions may be different in the two groups due to lack of blinding of NICU staff. However, the objective of the main trial is not to test the efficacy of ibuprofen for PDA closure (which has already been proven in previous placebo-controlled RCTs).8 Rather we intend to pragmatically test the effectiveness of an early selective treatment approach in improving clinical outcomes while allowing usual NICU care. If the larger RCT shows that similar patient-important clinical outcomes can be achieved with usual supportive NICU care without ibuprofen exposure even in extremely preterm infants with a symptomatic PDA, this will help to minimise early exposure of infants to these harmful medications; (2) Variation in echo diagnosis: Standardised echo assessments (online supplemental appendix B) are used across participating centres to minimise diagnostic variation.
Ethics and dissemination
The study has been approved by the IWK Research Ethics Board (#1027298) and six additional participating sites at the time of publication of this protocol.
Our integrated knowledge translation (KT) and end-of-study KT objectives and strategies are outlined as follows:
Integrated KT: (a) involvement of stakeholders in protocol development by organising regular virtual meetings before proposal submission for grant and ethics applications and (b) engagement of local site investigators for protocol adherence by organising quarterly virtual meetings and by development and dissemination of infographics of the study protocol to all participating sites.
End of study KT: (a) dissemination of results to wider Canadian and international neonatal community by presenting results at the Canadian Paediatric Society and Paediatric Academic Society’s Annual Meeting. The primary objective of end-of-grant KT will be to seek interest in the larger definitive trial from all stakeholders including clinicians and parent groups; (b) publication of results in peer-reviewed journals; (c) updating existing Cochrane review with trial results; (d) develop steering committee for the larger trial and involve parent representatives for outcome prioritisation; and (e) seek funding opportunities for the main RCT.
Discussion
PDA management remains one of the most controversial topics in neonatal intensive care with polarised opinions regarding treatment despite over 80 RCTs on this topic. This is likely attributable to two major limitations of existing RCTs: (a) lack of representation of the most vulnerable preterm infants at the highest risk of PDA attributable morbidity and (b) limitations in current definitions of haemodynamically significant PDA.
Most previous trials have included mature infants, with a mean GA of>26 weeks in 97% trials. Interestingly, a follow-up analysis of eligible infants who were not enrolled in the recently published PDA-TOLERATE trial due to the lack of physician equipoise showed that the group treated before 6 days postnatal age had a significantly lower incidence of CLD and CLD/death despite having lower GA, less receipt of antenatal steroids and substantially higher respiratory morbidity.36 These findings suggest that exposure to a moderate-large PDA shunt for ≥1 week in an extremely preterm infant could lead to adverse clinical outcomes such as CLD or death, irrespective of later PDA treatment. Further, the eligibility criteria with respect to hs-PDA definition have been wide, thus creating substantial heterogeneity in existing RCTs. A PDA size of >1.5 mm and the left atrium to the aortic root (LA:Ao) ratio of >1.4 have been the two most commonly used measures to define haemodynamic significance in RCTs.7 37 The major problem with this approach is that it does not differentiate between a moderate and a severe PDA shunt and completely ignores the clinical effects of the shunt volume. Martins et al demonstrated that the PDA size itself is weakly correlated to shunt volume.20 Furthermore, a PDA of a particular size may have a variable haemodynamic effect based on the infant’s pulmonary mechanics. This could possibly explain why the Beneductus trial, which enrolled preterm infants born at <28 weeks of GA with a left-to-right shunting PDA of any size of >1.5 mm, failed to demonstrate the benefits of early pharmacotherapy despite enrolling the smallest and sickest patients.13 Clyman et al, in a secondary analysis of their PDA-TOLERATE trial, demonstrated that prolonged exposure to a moderate-large PDA shunt (≥ 11 days) was associated with an increased risk of CLD, only in infants who received prolonged mechanical ventilation (≥10 days).38 Similarly, a secondary analysis of the TRIOCAPI trial also showed that moderate-large PDAs were associated with an increased risk of death or CLD only when infants required intubation for more than 10 days.39 Therefore, it is important to consider the clinical effects of PDA shunt in addition to echocardiography-confirmed markers of a large shunt volume on the preterm infant to identify PDA shunts that might benefit from closure.
The SMART-PDA trial attempts to address both the above-mentioned gaps in knowledge by exclusively enrolling the smallest preterm infants born at <26 weeks of GA and incorporating both clinical and echocardiographic criteria in its early treatment strategy. The proposed pilot trial will provide us with a valuable opportunity to explore the feasibility of conducting such a trial on a larger scale. The data generated from this pilot trial may further help us refine the highest risk population and, if required, incorporate prognostic or predictive enrichment strategies in the design of the definitive trial to decrease heterogeneity and improve the statistical power to detect a clinical meaningful effect within a reasonable timeframe.
Conclusion
There is limited evidence on whether, in the extremely preterm infants with a PDA, a selective early treatment strategy based on clinical and echocardiographic markers of moderate/severe shunt volume versus no treatment in the first postnatal week improves clinical outcomes. The SMART-PDA pilot trial will provide an opportunity to explore if such a trial is feasible in extremely preterm infants born at <26 weeks of GA who remain at high risk of adverse clinical outcomes despite recent advances in neonatal intensive care.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to acknowledge the contribution of late Dr John P Cleary from CHOC Children‘s, Orange County, USA, who was instrumental in guiding the development of this trial protocol. He was a neonatologist, a physician leader and mentor for many in the field of neonatal haemodynamics. He will be dearly missed by the neonatal community.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
X @souvik_neo, @santokh_Cardio, @DrLaley, @ajcammie
Contributors SM conceived the project, is the Principal Applicant on all related research grants, drafted the manuscript and is responsible for the overall content as guarantor. AHé provided expert input in the development of the project and reviewed the manuscript draft. MC provided expert input in the development of the project and reviewed the manuscript draft. TD drafted the statistical analysis section of the manuscript. WE-N provided expert input in the development of the project and reviewed the manuscript draft. SD provided expert input in the development of the echocardiography protocol and reviewed the manuscript draft. ZA provided expert input in the development of the project and reviewed the manuscript draft. JK provided expert input in the development of the project and reviewed the manuscript draft. ACK provided expert input in the development of the project and reviewed the manuscript draft. AHy provided expert input in the development of the project and reviewed the manuscript draft. KK provided expert input in the development of the project and reviewed the manuscript draft. MM provided expert input in the development of the project and reviewed the manuscript draft. DEW provided expert input in the development of the project, reviewed the manuscript draft. AJ provided expert input in the development of the project and reviewed the manuscript draft. FB is a patient partner who provided input on outcome prioritisation for the trial and reviewed the manuscript draft. AC was the primary research coordinator involved in trial oversight and day-to-day trial operations until July 2023 and reviewed the manuscript draft. TH is currently the primary research coordinator involved in trial oversight and day-to-day trial operations and reviewed the manuscript draft. JD provided expert input in the development of the project and reviewed the manuscript draft. PJM provided expert input in the development of the project and reviewed the manuscript draft. LT provided expert input on the statistical analysis section of the manuscript and reviewed the manuscript draft.
Funding This work was supported by the Canadian Institutes of Health Research (CIHR) Early Career Investigators in Maternal, Reproductive, Child and Youth Health Grant 2020 (#459750) with 1:1 matching fund support from the Dalhousie Medical Research Foundation (#001-A); IWK Health Research (#1025843); Department of Pediatrics, Dalhousie University (RN446947-459750). The funding agencies have no authority over study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, conduct, reporting or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.