Article Text

Abstract
Introduction Premature birth and very low birth weight (VLBW) are leading causes of neonatal mortality. Almost all premature infants experience hyperbilirubinaemia. Administering probiotics to breastfeeding mothers may positively affect infant outcomes. This trial aims to investigate whether probiotic supplementation for mothers with VLBW infants affects total serum bilirubin levels and postpartum depression scores (primary outcomes), as well as some other neonatal and maternal outcomes (secondary outcomes).
Methods and analysis This is a randomised, double-blind, placebo-controlled superiority trial with two parallel arms. Participants, caregivers and outcome assessors will be blinded. A total of 122 breastfeeding mothers of neonates with a birth weight of 1000–1500 g, along with their infants within 48 hours of birth, will be assigned to either the probiotic or placebo group in a 1:1 ratio through block randomisation, stratified by singleton and twin births. The intervention will involve oral administration of probiotics containing Lactobacillus paracasei 431 and Bifidobacterium lactis BB-12, or an indistinguishable placebo, for 42–45 days. Outcomes will be assessed through daily observations, laboratory assessments and the Edinburgh Postpartum Depression Scale. Adverse events will also be documented. Modified intention-to-treat analyses will be employed for the primary and secondary outcomes, excluding participants lost to follow-up from all postintervention assessments.
Ethics and dissemination This study protocol has been approved by the Medical University of Tabriz Ethics Committee (IR.TBZMED.REC.1401.735). Findings will be disseminated through publication in a peer-reviewed journal and presentations at relevant conferences.
Trial registration number IRCT20100414003706N42.
- Clinical Trial
- Neonatal intensive & critical care
- Depression & mood disorders
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
A low risk of selection bias due to the appropriate random assignment of participants to study groups and allocation concealment.
A low risk of performance and detection biases due to the implementation of blinding for participants, care providers and outcome assessors.
Conducting the trial in a tertiary hospital that covers very low birthweight infants from diverse geographical areas enhances the generalisability of the study results.
A lack of long-term assessment of intervention effects, primarily due to financial, time and logistical constraints.
The omission of biological collections from the protocol due to financial constraints limits the understanding of the probiotics’ effects on the maternal and neonatal microbiome.
Introduction
Premature birth and very low birth weight (VLBW, defined as weighing less than 1500 g) are the leading causes of infant mortality.1 About 60% of infant deaths are attributed to VLBW. In Iran, infants weighing between 1000 and 1500 g have shown survival rates of 45% on discharge and 58% at 28 days after birth.2 Over the past three decades, advancements in perinatal and neonatal intensive care, such as the utilisation of antenatal steroids, surfactants and innovative mechanical ventilation therapies, have markedly enhanced the survival rates of VLBW infants.3–5 Despite these improvements, complications like sepsis, necrotising enterocolitis, bronchopulmonary dysplasia, intraventricular haemorrhage and retinopathy of prematurity are still common among these vulnerable infants.6 7
Jaundice affects nearly 80% of premature neonates within the first week of life.8 Despite the progress made in the treatment of hyperbilirubinaemia, it remains a major cause of neonatal morbidity and mortality.9 The current approach to managing hyperbilirubinaemia in premature neonates focuses on determining the age-specific bilirubin threshold necessitating the initiation of phototherapy. However, concerns exist regarding the potential adverse effects of invasive phototherapy in premature neonates.10 In addition to causing stress for parents, phototherapy impedes breastfeeding and contributes to other problems, such as fluctuating neonate body temperature and dehydration. Therefore, complementary approaches may offer valuable support in this context.11
Pregnancy and childbirth are transitional periods in women’s lives. However, they can lead to permanent and long-term depression, with an estimated 9–16% of women experiencing postpartum depression (PPD).12 The prevalence of PPD in Iran is reported to be approximately 25% (95% CI 23% to 28%).13 The results of a recent systematic review have shown that preterm birth increases the risk of PPD in women by about 29%.14 However, most women with PPD lack access to psychotherapeutic interventions or are unwilling to take antidepressants. Thus, there is a pressing need for safe and effective preventive and therapeutic interventions to address this critical issue.15
Probiotics are live microorganisms that resemble those found in the human gut and are known to confer health benefits when administered in appropriate doses. Studies examining the safety of probiotics have not revealed any significant adverse effects on breastfeeding mothers and their infants.16 17
When administered to breastfeeding mothers of VLBW infants, probiotics may affect bacterial colonisation of the infants.18 According to the entero-mammary pathway theory, beneficial intestinal bacteria are transferred to the mammary glands via dendritic cells during late pregnancy and breastfeeding.19 20 Moreover, probiotics can affect the concentration of bilirubin in the liver-intestinal circulation by inhibiting the degradation of conjugated bilirubin, as well as by enhancing intestinal peristalsis and defecation, thereby increasing bilirubin excretion.16 21 In addition, probiotics may influence mothers’ mental health by reducing stress and depression.22
The Cochrane systematic review,18 published in 2018, found only one trial on 49 mothers of 58 infants concerning the effect of postnatal probiotic administration to breastfeeding women on the morbidity of VLBW neonates, in which the effect on hyperbilirubinaemia was not studied.23 The review authors emphasised the need for further trials in the field. Although our recent trial in this field24 showed some promising results, its sample size was limited (25 in each group) and the administered probiotics contained only one strain (Lacticaseibacillus paracasei subsp paracasei, 1.5×109 colony-forming units (CFU)/day), and it did not assess maternal mental health outcomes.
A recent systematic review (2022) identified only two studies examining the effect of probiotic administration from pregnancy through the postpartum period on PPD. The meta-analysis from these studies revealed no statistically significant difference in women’s depression between the probiotic and control groups. This indicates a lack of evidence supporting the effect of probiotics on PPD, emphasising the need for further investigation into their effect on maternal mental health.25
To the best of our knowledge, the present study represents the first trial with a relatively large sample size, aiming to investigate the effects of orally administering a combination of two probiotic strains to breastfeeding mothers of VLBW neonates on neonatal outcomes. Additionally, it is one of the few trials assessing the intervention’s effect on PPD without considering the administering period, and it is the first trial assessing the effect of postpartum administration on PPD.
Methods and analysis
Study objectives
This trial aims to assess the effects of orally administering probiotics containing L. paracasei 431 and Bifidobacterium lactis BB-12 at a dose of 1×109 CFU/day each to breastfeeding mothers of neonates with VLBW on total serum bilirubin (TSB) levels and PPD scores, which are the primary outcomes. Additionally, we will assess the effects of the supplementation on rate of phototherapy, any serious complications, and feeding intolerance on the 7th day of intervention, as well as weight gain, length of and duration of total parenteral nutrition (TPN) as infant secondary outcomes. Also, mastitis and postpartum anxiety were assessed as maternal secondary outcomes.
Hypotheses
Supplementing breastfeeding mothers with L. casei 431 and B. lactis BB-12 probiotics reduces the TSB levels in VLBW infants by a minimum of 25%.
Supplementing breastfeeding mothers with L. casei 431 and B. lactis BB-12 probiotics reduces PPD symptom scores by at least 30%.
Study design and setting
This is a randomised, double-blind, placebo-controlled trial with two parallel arms aiming for superiority. The study will take place at Al-Zahra Educational Hospital in Tabriz, Iran, which serves as the primary referral centre in northwest Iran for VLBW infant deliveries and hospitalisation of VLBW infants born in the other hospitals. Participant recruitment commenced on 30 December 2022, and the anticipated completion date is October 2023.
Participants
Participants include breastfeeding women and their VLBW infants, whether singletons or twins. In the case of twin infants, both will be included if eligible.
Inclusion criteria
Infants with a birth weight between 1000 and 1500 g who can receive breast milk.
Delivery of a singleton or twins within the past 48 hours.
Women who desire and are capable of breastfeeding their babies and can be present at the hospital where the baby is admitted at least once a week.
Infants hospitalised for at least 7 days after the intervention commences.
Exclusion criteria
Contraindication to breastfeeding.
Obvious anomalies and/or poor conditions in infants diagnosed by a neonatologist.
Known serious illnesses in women.
Regular use of probiotics (in any form) by the woman.
History of an allergy to probiotics.
Immunodeficiency in the mother and/or infant(s).
Procedure
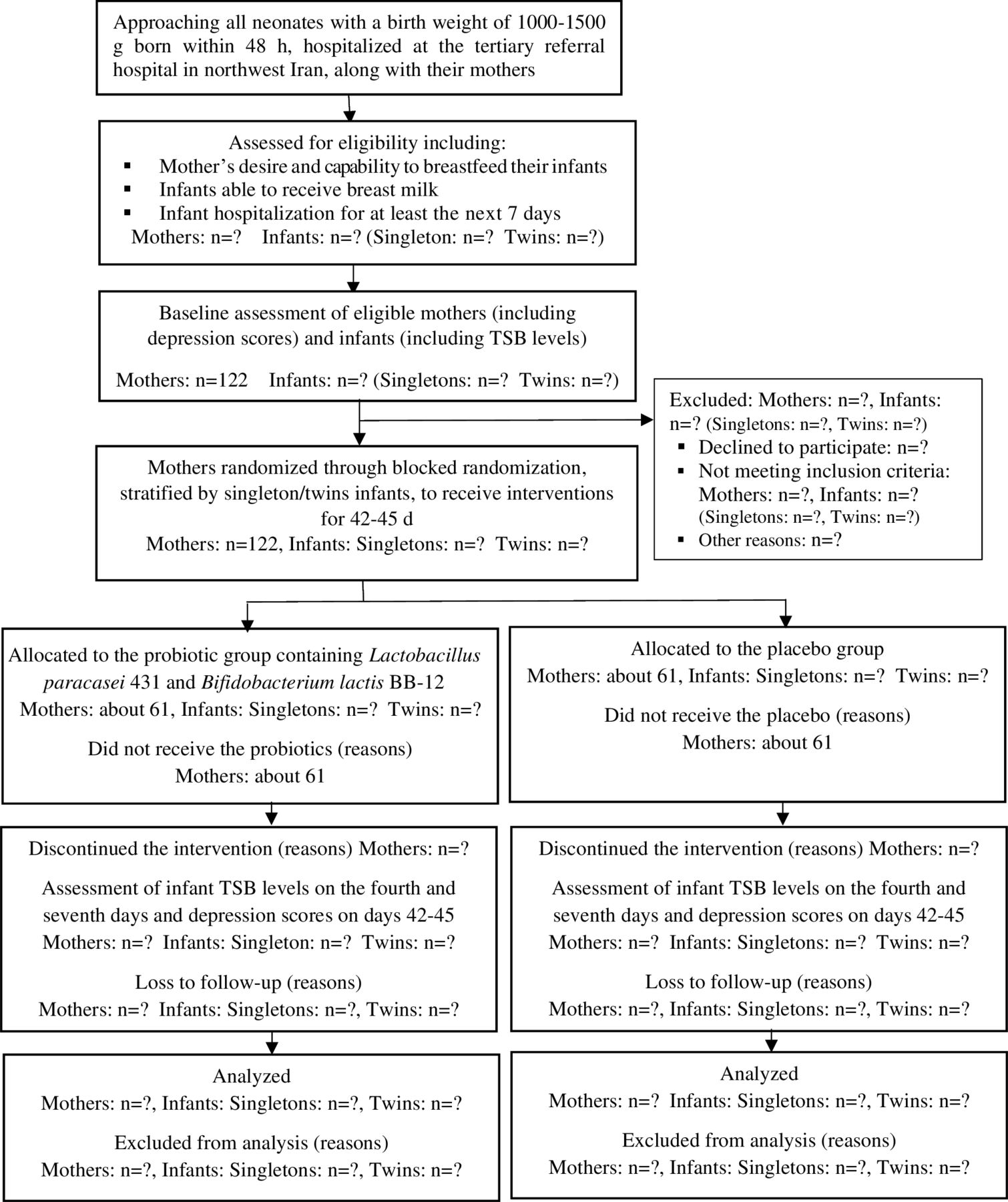
The principal investigator (PI; first author, MA) will explain the study aims and procedures to the mothers (and, if possible, to their husbands or another legal guardian), assess the eligibility criteria and ask them to sign written informed consent forms (see online supplemental material). Women will also be informed that they can leave the study at any time without providing a reason. After obtaining informed consent and collecting baseline data, the participants will be randomised into one of two groups: maternal probiotic supplementation or control (figure 1 presents a flow chart of the trial process).
Supplemental material

{kind=link}
Flow chart of the trial process. TSB, total serum bilirubin.
Randomisation and blinding
The allocation sequence will be generated using block randomisation, with randomly varying block sizes of 4 and 6 and an allocation ratio of 1:1 in each stratum referring to the random.org computer program. Stratification will be based on whether the infants are singletons or twins. Identical, opaque and sequentially numbered (within each stratum) bottles containing identical capsules (probiotic/placebo) will be used to conceal the sequence and to maintain blinding. Participants within each stratum will receive the bottles in the sequence corresponding to their enrolment in the study. Trial participants, care providers, outcome assessors and data analysts will be blinded. The allocation sequence will be generated and the bottles prepared by a person not involved in participant recruitment, allocation or data collection. Unblinding is permissible in the unlikely event of an adverse occurrence. In such a case, the study coordinator who generated the allocation sequence should inform the neonatologist colleague (MMG, a highly experienced faculty member) of the participant’s intervention assignment.
Intervention
Participants will receive either probiotics or a placebo for 42–45 days. One package of L. paracasei 431 and one package of B. lactis BB-12 at a concentration of 1012 CFU/g each were sourced from ‘Hansen Co., Denmark’. Corn starch was acquired from ‘Glucose Co, Iran’. Afterwards, 2.56 g of each strain will be mixed with 1281 g of corn starch in the laboratory of the pharmacy department at Tabriz University of Medical Sciences and filled into 2562 capsules (each containing 500 mg) as a probiotic supplement. Additionally, 2562 capsules will be filled with corn starch to serve as the placebo. This probiotic is lyophilised, meaning that it is slowly ground in a sterile mortar to produce very fine and uniform particles, which are then mixed with a filler to ensure that 500 mg contains 1×109 probiotic bacteria per strain. Before filling the capsules, the powder will be sampled and cultured for microorganisms. Microbial culture and counting will also be performed on the sample of the filled capsules to ensure quality.
To ensure consumption, capsules will be provided to breastfeeding mothers daily while their infants are in the hospital. If daily access to the mothers is not feasible, the packages will be delivered, with no more than one package per week. If an infant is discharged before the 42–45 days of intervention period, all remaining capsules will be provided to the women and they will be instructed on home supplementation and contacted daily for follow-up. All capsules are taken and any adverse events will be recorded in a diary. Participants will be instructed to take one capsule daily, preferably before or during a meal, store the capsules in a cool, dry place (below 25ᵒC) and take the supplement at least 1 hour before or after other supplements.
The newborns’ TSB levels will be measured at baseline, after obtaining informed consent, as well as on days 4 and 7 after intervention at the laboratory of Al-Zahra Educational Hospital. To ensure result validity, 10 blood samples will be sent for testing both to the comprehensive reference laboratory in Tabriz and to the hospital laboratory before the study commences, to establish the intraclass correlation coefficient (ICC).
A checklist has been developed to meticulously document the commencement and cessation of each phototherapy type and any blood exchange transfusions administered during the 42–45 days following the intervention. In initiating and discontinuing phototherapy, the neonatologists at the hospital follow the Queensland Maternity and Neonatal Clinical Guidelines while consulting the Plot TSB levels on the nomogram, considering gestational age, weight and age appropriateness. Phototherapy is discontinued once the bilirubin level lowers to a safe threshold based on the guidelines.26 In severe cases with bilirubin levels exceeding 25–30 mg/dL, a blood exchange transfusion is performed to prevent kernicterus.27 The PI will complete the checklist, also document the occurrence of any significant neonatal issues such as sepsis and necrotising enterocolitis daily based on the infant’s medical record and confirmation of the neonatologist colleague. The infant’s anthropometric measurements (weight, height and head circumference) at birth, on the seventh day after the intervention and at discharge will be recorded based on the infant’s medical record. The infant’s weight will be measured using a calibrated scale in the neonatal intensive care unit (NICU) department. At the 42–45 days of in-person meeting, the PI will record the infant’s anthropometric characteristics, rehospitalisation and phototherapy needs after discharge, infant mortality, other neonatal problems, and maternal health (assessed using the Edinburgh Postnatal Depression, Anxiety and Mastitis Scales), and any side effects. Both study groups will receive standard care provided by health centres and the hospital. Table 1 shows the schedule of enrolment, interventions and assessments.
Schedule of enrolment, interventions and assessments
Follow-up
Follow-up assessments will occur daily over the period of 42–45 days, either in person or via phone calls, depending on the infant’s hospitalisation status and the woman’s condition. The women will be encouraged to attend the hospital daily during their infant hospitalisation for capsule consumption and maternal mastitis symptom assessment. Any adverse events during the study period will be documented. Serious adverse events will be promptly addressed by a specialist. Mothers observing any adverse events in themselves or their infants will call the PI, who will arrange for a specialist or neonatologist visit at the hospital, free of charge.
Outcomes
Primary outcomes are the TSB levels on the fourth and seventh days following the intervention, as well as the depression score at 6 weeks post partum.
Secondary infant outcomes include the duration of phototherapy, the infant’s weight on the seventh day and the 42–45 days of postintervention, the time taken to achieve full oral feeding, a composite variable of occurrence of serious neonatal problems (such as sepsis, necrotising enterocolitis, bronchopulmonary dysplasia, retinopathy of prematurity and intraventricular haemorrhage) up to 42–45 days of infancy and duration of infant hospitalisation.
Secondary maternal outcomes include the occurrence of mastitis during the 42–45 days of intervention period and the anxiety score at 6 weeks post partum.
Adverse events
Reported adverse events of probiotics include sepsis, vomiting, loose stools, abdominal distension and bloating. These events will be monitored daily throughout the 42–45 days of intervention period using a diary. Any additional adverse events occurring within this period will also be recorded in the diary. In the event of severe complications during the study leading to significant discomfort, treatment allocation will be promptly unblinded to the specialist and/or the person in charge in the ward if requested, to facilitate the immediate provision of necessary support and treatment, provided free of charge under the direct supervision of a designated perinatologist or neonatologist from the research team. To maintain transparency and accountability, any unblinding due to severe complications will be promptly reported to the research ethics committee. This communication serves to keep the committee informed of significant events and enables them to offer guidance or take necessary actions to protect the interests of all participants.
Sample size
The sample size was calculated using G*Power software based on the TSB level and PPD score as the primary outcomes. Referring to Matin et al’s study24 regarding the TSB level on the seventh day after intervention, with parameters m1=6.0 mg/dL, m2=4.5 mg/dL (assuming a 25% reduction due to the intervention), SD1=SD2=2.3, two-sided alpha=0.05 and 90% power, the sample size was calculated to be 51 individuals per group. Additionally, based on Vaziri et al’s study28 regarding depression, with parameters m1=7.18, m2=5.03 (assuming a 30% reduction due to the intervention), SD1=SD2=3.99, two-sided alpha=0.05 and 80% power, the sample size was calculated as 55 per group. Therefore, due to the larger sample size calculated based on the PPD variable and accounting for a 10% attrition rate, this study will include 61 women in each group, totalling 122 breastfeeding mothers with VLBW infants and their eligible infants.
Interim analysis
There will be no interim analysis conducted in this study.
Data collection
All data will be collected by the PI, a doctoral candidate in midwifery who has received thorough training from supervisors, including the neonatologist on the study team. The neonatologist, a highly experienced faculty member who is present in the study setting every working day, will directly oversee the data collection process. The following tools will be used for data collection:
Maternal baseline assessment questionnaire: It includes information on the mother’s education level, date of birth, gestational age at delivery, number of pregnancies, number of abortions, number of live children, number of stillbirths, number of child deaths after birth, history of caesarean section, number of prenatal care visits, pre-pregnancy complications (such as thyroid disorder, chronic hypertension, pre-eclampsia), smoking and drug use during pregnancy, spouse’s smoking, complications during pregnancy (such as vaginal bleeding, placental abruption, amniotic sac rupture, oligohydramnios, polyhydramnios) and predelivery magnesium sulfate usage and corticosteroid intake. The PI will complete the questionnaire through interviews with the participants and by reviewing her medical records after enrolment.
Characteristics of hospitalised infant questionnaire: TSB levels will be measured using a venous blood sample at baseline and on the fourth and seventh days after intervention. Other data which will be recorded include infant’s hospitalisation date, blood group, gender, first and fifth-minute Apgar score, birth weight, height and head circumference at birth, nutrition status assessment during the first day of enrolment, surfactant usage, continuous positive airway pressure use, need for phototherapy, duration and type of phototherapy, infant death, age at death, the time that the infant can tolerate more than half of the oral feeding with breast milk, the time that the infant can tolerate 100% oral feeding with breast milk, cessation of intravenous feeding (TPN), duration of antibiotic treatment, neonatal complications (such as necrotising enterocolitis, intraventricular haemorrhage, sepsis, positive blood culture for sepsis, pulmonary dysplasia), examination on the seventh day of the intervention (including weight, height, head circumference, nutritional status), assessments on the seventh day and at discharge (infant’s age, weight, height, head circumference and nutritional status) and assessments on the 40th–45th days (including infant’s death, age at death, weight, height, head circumference, retinopathy of prematurity, blood transfusion, rehospitalisation, type of feeding in the last 24 hours). The PI will collect these data via neonate medical records, interviewing the corresponding neonatologist, consulting the neonatologist colleague and interviewing the mothers.
The Edinburgh Postnatal Depression Scale (EPDS): The enrolled mothers will complete the scale at baseline (before the intervention) and 42–45 days after delivery. The EPDS was developed by Cox et al in 1987 and is used to measure depression during pregnancy and after delivery. This scale consists of 10 items with four options. The options for each item are assigned a score from 0 to 3 based on severity of the symptom, and the total score is obtained from the sum of the scores, which can vary from 0 to 30. The concurrent correlation coefficient of the original version of the scale with the Beck Depression Scale was 0.78. The reliability of this scale was estimated at 0.75 using Cronbach’s alpha and the split-half method.29 Its Persian version has been validated in Iran, and its internal consistency using Cronbach’s alpha ranges from 0.77 to 0.86.30
The Spielberger State Anxiety Scale (SSAS) will be administered at baseline, and the Postpartum Specific Anxiety Scale Research Short-Form (PSAS-RSF) will be administered on the 40th–45th days after delivery. The SSAS assesses state and trait anxiety; however, only state anxiety will be assessed in this study using 20 questions. The questions are arranged with four options (not at all, somewhat, moderate, very much), and the total scale ranges from 20 to 80 (20=no anxiety, 21–39=mild anxiety, 40–59=moderate anxiety, 60–79=severe anxiety, 80=very severe anxiety). This questionnaire demonstrates a high level of validity and reliability. The validity of its Persian version has been confirmed in Iran by Mahram, with an internal consistency of 0.91 using Cronbach’s alpha.31 The initial version of the PSAS includes 51 items.32 However, the 16-item version of the PSAS-RSF is considered the strongest version in terms of theory and psychometrics. The PSAS-RSF is the first validated short form specifically designed to measure postpartum anxiety. This scale can be used for up to 12 months after delivery. Each item is scored on a ‘Four-State Ordinal Continuum’ between 1 (never) and 4 (always), and the total scale score ranges from 16 to 64. The scale has demonstrated good reliability (McDonald’s ω=0.88) for the entire instrument.33 The Persian version of this scale has been validated in Tabriz, Iran, with high internal consistency (Cronbach’s alpha 0.72) and test–retest reliability (ICC 0.97 (95% CI 0.98 to 0.93)).34
A mastitis checklist will be used to assess signs and symptoms in the breasts (redness, pain, tenderness and existence of a hard mass), as well as influenza (fatigue, fever, shivering or chills). If the woman reports at least two symptoms of breast and one symptom of influenza, a diagnosis of mastitis will be made.
The use of probiotic/placebo supplements and any adverse events will be recorded daily by the mothers in a diary.
Content validity of the tools used in this study (except the validated ones) will be determined using the opinions of experts, including obstetric specialists, neonatologists, midwives and nurses.
All collected data will be transparently reported to an independent auditor assigned by the Ethics Committee of Tabriz University of Medical Science whenever it is requested or necessary.
Data management
Data entry will occur in the software immediately after data collection. To ensure accuracy, range checks will be implemented for the data values. Additionally, the data from the first five participants and approximately 10% of the remaining participants (randomly selected) will undergo a thorough review by another individual.
Regular reminders will be sent to enhance both protocol adherence and participant retention. All required data will be collected from all participants, including those who are non-adherents. Reasons for non-adherence and non-retention will be inquired directly from participants or, if a participant is inaccessible, from a designated support person. The reasons will be reported by the study group. In the informed consent form, participants’ consent will be obtained for the use of their and their babies’ electronic and paper medical records, which may facilitate the collection of some important data from those lost to follow-up.
Confidentiality
To maintain complete confidentiality, we will not include the identifiable data from potential and enrolled participants in their questionnaires or in the computer software where data are entered. Participants will be identified by unique codes. Identifiable participant characteristics, along with their corresponding codes, will be stored separately in a secure location accessible only to the data collector, corresponding author and the auditor assigned by the ethics committee. Under specific circumstances, other members of research team and ethics committee representatives can access these details with a valid rationale.
Statistical analysis
Modified intention-to-treat analyses will be conducted for the primary and secondary outcomes, excluding those lost to follow-up from all postintervention assessments of the outcomes.
The normal distribution of quantitative outcomes across groups will be examined using the Kolmogorov-Smirnov test. If the data are normally distributed, repeated measures analysis of variance will be employed to compare the groups concerning TSB levels assessed on the fourth and seventh postintervention days, adjusting for the baseline values. The interaction effect of group and time will also be evaluated. Analysis of covariance will be used to compare the other quantitative outcomes, such as PPD and anxiety scores assessed once after intervention, adjusting for baseline values.
Assumptions, such as sphericity and the absence of spurious outliers, will be verified before interpreting the results. Non-parametric tests, such as the Mann-Whitney U test, will be used if parametric assumptions are not met. Binary logistic regression will be used to compare the groups regarding qualitative outcomes, such as the need for phototherapy, infant food tolerance, the occurrence of important neonatal problems and maternal mastitis. A p value level of <0.05 will be considered statistically significant, and all analyses will be conducted using IBM SPSS (V.24).
Trial results will be disseminated through publication in peer-reviewed journals and presentations at professional society meetings.
Patient and public involvement
Patients and the public were not involved in the design of the study or the recruitment of participants. At the end of the study, the main results will be disseminated to participants through peer-reviewed journals and presented at international conferences.
Ethics and dissemination
This study protocol has been approved by the Medical University of Tabriz Ethics Committee (IR.TBZMED.REC.1401.735). Participants will give informed consent to participate in the study before taking part. Any protocol amendments will be notified to the ethics committee and the Iranian Registry of Clinical Trials. Findings will be disseminated through publication in a peer-reviewed journal and presentations at relevant conferences.
Discussion
Low birth weight is one of the most common contributors to increased physiological bilirubin levels in infants. Elevated bilirubin production in premature infants increases the risk of mortality and long-term neurodevelopmental impairment due to bilirubin neurotoxicity.35 36 VLBW infants are particularly vulnerable to brain damage from hyperbilirubinaemia and generally receive treatment at lower thresholds than normal birthweight infants.37 Two common treatments for neonatal hyperbilirubinaemia (ie, phototherapy and blood transfusion) are associated with diverse side effects.38 Therefore, considering the high prevalence of hyperbilirubinaemia and the importance of its prevention and rapid treatment in premature infants, it is essential to use alternative or supportive methods that have minimal or no side effects. The results of a systematic review and meta-analysis revealed evidence of the effect of probiotic supplements on reducing the duration of phototherapy, TSB at 96 hours and on the seventh day after birth, as well as the duration of hospitalisation in infants with hyperbilirubinaemia. However, the authors stated that the certainty of this evidence is low, and that further robust studies are necessary to confirm their efficacy.39 This trial seeks to provide evidence for such an alternative therapy, which may have other beneficial effects on infants and their mothers.
In our study, we will use two strains, L. paracasei 431 and B. lactis BB-12, well-studied strains with documented safety and effectiveness in various health aspects.40 41 Numerous clinical trials involving these strains have demonstrated their positive impact on gut microbiota, immune function and gastrointestinal health.42 43 L. paracasei 431 has been noted for its ability to modulate immune responses and enhance barrier function in the gut,44 while B. lactis BB-12 is known for its positive effects on microbial balance and its potential to reduce pathogenic bacteria.45 Our research team and others have successfully used these strains in previous studies, which provided us with a foundation of experience and data to build on.24 46–48 In this study, these strains were sourced from Chr Hansen and meet the necessary international standards. The formulations containing these strains are currently available in the Iranian market and have been approved by the Iranian Food and Drug Administration of the Ministry of Health and Medical Education. This approval ensures that the strains are safe, effective and manufactured according to good manufacturing practices. Products containing these strains, which can be found in pharmacies and health food stores in Iran, are often marketed for their benefits to digestive health and immune function.
There are no strict recommended guidelines for the dosage. However, the dosage of 1×109 CFU/day from each strain falls within the range commonly used in clinical trials and aligns with guidelines for probiotic administration. This dosage has been effective in various studies without causing adverse effects.49 Additionally, the promising results in our previous study, where only L. paracasei subsp paracasei at 1.5×109 CFU/day was used,24 provided the rationale for selecting the dosage in the current investigation.
Strengths and limitations
Recruiting participants within the first 48 hours after delivery in this study can make it possible to thoroughly investigate the preventive effect of probiotic supplements on hyperbilirubinaemia in VLBW infants. The primary outcome of infants’ TSB levels will be measured at baseline, as well as on the fourth and seventh days after intervention. This timeline aligns with the peak and resolution of neonatal hyperbilirubinaemia in the VLBW infant population.38 50
Adhering rigorously to clinical trial standards, including proper randomisation and allocation concealment, blinding of participants, care providers, and outcome assessors, comprehensive participant follow-up, strict adherence to the study protocol in its implementation and transparent reporting of all results, we aim to minimise the risk of bias such as selection, performance, detection, attrition and reporting biases. Also, conducting the trial in a tertiary hospital covering VLBW infants from diverse geographical areas enhances the generalisability of the study results.
Due to budgetary constraints, this trial does not incorporate the collection and analysis of biological samples such as blood, maternal stool, maternal milk or fetal stool, which could offer valuable insights into the probiotic effects on the maternal and neonatal microbiome. This omission limits our ability to fully understand the mechanisms behind any observed effects on neonatal and maternal outcomes. Future research should prioritise including microbiome analysis to enhance our comprehension of probiotic roles in maternal and neonatal health, particularly in the context of VLBW infants.
In this trial, we are going to assess the short-term intervention’s effects on a few maternal short-term outcomes. Thus, it may not fully capture the intervention’s impact on the overall well-being of women. Future studies should assess broader maternal outcomes, such as sleep quality, to provide a more holistic view of the probiotic intervention’s influence on maternal health.
The primary outcome of PPD scores will be assessed once after intervention, that is, at the end of the 42–45 days of intervention period, aligning with the conventional time frame for assessing PPD. This time frame allows for examination of the immediate impact of probiotic supplementation on maternal mental health.51 Assessing PPD at two time points (baseline and 42–45 days after delivery) may not capture fluctuations adequately, and more frequent evaluations, coupled with clinical assessments, could yield comprehensive results. However, due to the vulnerability of women with VLBW infants, frequent assessments could impose a burden on participants. Therefore, a more frequent assessment of PPD is recommended in future studies on less vulnerable women.
Since most VLBW infants admitted to the hospital from which we will be recruiting participants come from other cities, including other provinces, it may be challenging to retain participants in the long term after discharge due to limited physical access. However, previous studies have demonstrated the positive effects of short-term direct probiotic administration on newborns, including a reduction in the duration of phototherapy and TSB levels.39 In a trial, administering B. breve and L. rhamnosus orally at a concentration of 2×106 CFU/day, starting from the first hour of life, resulted in rapid and substantial colonisation by days 5 and 6.52 To our knowledge, no studies have investigated the effects of postpartum probiotic administration on PPD. Nevertheless, a trial has shown a positive effect from 4 weeks of probiotic supplementation on certain cognitive functions in patients with major depressive disorder.53 Therefore, based on the evidence supporting the effectiveness of short-term interventions and the practical challenges involved, our research team chose a period of 42–45 days of intervention. This duration strikes a balance between providing sufficient treatment time and addressing the logistical challenges of the study. Long-term interventions, coupled with extended assessments of maternal and infant outcomes in future studies, could provide deeper insights into the impacts of intervention.
Ethics statements
Patient consent for publication
Acknowledgments
We thank the Vice-Chancellor for Research at Tabriz University of Medical Sciences for their financial support.
References
Footnotes
Contributors MA, SM-A-C, MM, MMG, AH-R, ZF and MS contributed to the design of the protocol, critically read the paper, provided inputs and revisions and approved the final manuscript. SM-A-C and MA contributed to the implementation and analysis plan and have written the first draft of this paper. MS is the corresponding author of this article. SM-A-C is the guarantor.
Funding This study is funded by Tabriz University of Medical Sciences.
Disclaimer The funding source had no role in the design and conduct of the study or the decision on writing and submitting this manuscript.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.