Article Text

Abstract
Introduction Heart failure with preserved ejection fraction (HFpEF) is a prevalent comorbidity among patients with end-stage kidney disease. Although sodium-glucose cotransporter 2 inhibitors are validated in treating heart failure and ameliorating left ventricular hypertrophy among non-dialysis patients, the effects on dialysis patients are unknown. We previously investigated the pharmacokinetics of henagliflozin in patients undergoing haemodialysis (HD) or peritoneal dialysis (PD) and clarified its safety.
Methods and analysis This multicentre, randomised, double-blind, placebo-controlled trial is being conducted at three hospitals in Shanghai, China. A target of 108 HD or PD patients with HFpEF are randomly allocated to treatment group (henagliflozin 5 mg/day in addition to standard therapy) or control group (placebo with standard therapy) at a ratio of 1:1. All subjects will be followed up for 24 weeks. The primary outcome is change in echocardiography-measured left ventricular mass index. The secondary interests include changes in left atrial volume index, E/e’, e’ and N-terminal pro-B-type natriuretic peptide (NT-proBNP). Intergroup comparisons of change in echocardiography-related outcomes from baseline to 24 weeks are based on a linear regression model adjusted for baseline values (analysis of covariance), and repeated measure analysis of variance with Bonferroni adjustment is employed for comparison of change in NT-proBNP. Subgroup analyses of the primary and secondary outcomes are conducted to determine whether the effect of henagliflozin varies according to dialysis modality. The χ2 method is used to compare the occurrence of adverse events and severe adverse events.
Ethics and dissemination This trial has been approved by the Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University (LY2023-127-B). All participants provide written informed consent before screening. The results of the trial will be disclosed completely in international peer-reviewed journals. Both positive and negative results will be reported.
Trial registration number ChiCTR2300073169.
- Dialysis
- Heart failure
- Randomized Controlled Trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is a multicentre, randomised, double-blind, placebo-controlled trial in dialysis patients that addresses the complication of heart failure with preserved ejection fraction.
It provides important 24-week data about the effects of sodium-glucose cotransporter 2 (SGLT2) inhibitor on cardiac geometric and functional alterations and about the safety of the treatment.
The trial includes treatment group with henagliflozin and control group with placebo, and the primary outcome is echocardiography-measured left ventricular mass index.
Since mortality and new-onset cardiovascular diseases are not the designated outcomes, the trial cannot clarify whether SGLT2 inhibitor reduces the risk of hard endpoints.
Physical tests objectively evaluating cardiac function, for example, 6 min walk test, are absent in the trial.
Introduction
Background
Chronic kidney disease (CKD) is a growing public health priority. A significant proportion of patients with CKD progress to end-stage kidney disease (ESKD) requiring renal replacement therapy. In 2017, the prevalence of CKD was estimated at up to 9.1% of the global population and the number of patients undergoing dialysis was more than 3.14 million across the world.1 Cardiovascular death is 10-fold to 100-fold greater among patients with ESKD than in age-matched and sex-matched people of the general population without kidney disease.2 In contrast to the general population, heart failure (HF) is a more important cause of cardiovascular mortality in dialysis patients instead of myocardial infarction or stroke.3
HF as a common comorbidity at the initiation of dialysis presents in 36% of patients with ESKD,4 and the incidence of de novo HF during dialysis therapy is about 7% per year.5 Heart failure with preserved ejection fraction (HFpEF) is the most common phenotype in patients on dialysis, accounting for half of the cases.6 Left ventricular hypertrophy (LVH) is the predominant pathophysiological mechanism in HFpEF, and an elevated left ventricular mass (LVM) portends an increased risk of death.7
The widely accepted strategies to treat HF in dialysis patients consist of restriction of sodium and fluid intake, preservation of residual kidney function (RKF), sufficient ultrafiltration, and appropriate treatment for hypertension, hyperglycaemia, anaemia, etc. Medications including renin–angiotensin system inhibitors, angiotensin receptor-neprilysin inhibitors (ARNI), β-blockers, mineralocorticoid receptor antagonists (MRA) and loop diuretics in the presence of RKF are recommended.8 9 However, there has been no proven therapy for HFpEF in the setting of dialysis,10 resulting in excessive mortality, increased hospitalisation, impaired quality of life and aggravating health burden. Novel treatment is warranted for this population.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are a relatively new class of antidiabetic agents that exert beneficial effects on kidney and cardiac outcomes.11 For non-dialysis population, SGLT2 inhibitors are recommended for HF as a new treatment, in addition to conventional therapies, irrespective of the presence of type 2 diabetes and the phenotypes based on the measurement of left ventricular ejection fraction (LVEF), and the cardiac protective role is endorsed by the latest guidelines.12–14
An animal study revealed that 2-month treatment of empagliflozin regressed LVH in an HF model of pigs, as demonstrated by cardiac magnetic resonance (cMR), echocardiography and histology.15 As supported by a meta-analysis, all interventions modifying left ventricular (LV) remodelling, including LVH in the short term, improve patient survival in the long term.16 Recent meta-analysis concluded that in non-dialysis populations, SGLT2 inhibitors may be associated with improvement in LVM and E/e’.17 18 Therefore, it is possible that the cardioprotective effect of SGLT2 inhibitors is mediated via improvement in LV geometry.
The exact mechanisms of cardiac protection by SGLT2 inhibitors have not been clearly elucidated yet, and it may ameliorate LVH and HF through off-target effects.19 Hence, patients on dialysis with HFpEF may benefit from SGLT2 inhibitors theoretically. As presented by the EMPA-KIDNEY and DAPA-CKD trials, SGLT2 inhibitors can lower the risks of kidney disease progression or cardiovascular death despite impaired kidney function in which an estimated glomerular filtration rate (eGFR) is less than 45 mL/min/1.73 m2 or 30 mL/min/1.73 m2.20 21 However, due to a lack of safety and efficacy data, SGLT2 inhibitors are not validated in those with an eGFR below 20 mL/min/1.73 m2, and dialysis is generally listed as a contraindication to administration.
Rationale
Henagliflozin is an oral selective inhibitor of SGLT2 approved by the State Food and Drug Administration in China for type 2 diabetes mellitus with eGFR at least 30 mL/min/1.73 m2, with a recommended dose of 5–10 mg daily.
We conducted a randomised, open-label study to explore the pharmacokinetics of henagliflozin in haemodialysis (HD) or peritoneal dialysis (PD) patients with diabetes22 and compared the results with those from a previous study in patients with diabetes with normal kidney function.23 When treated with a single dose of 5 mg henagliflozin, dialysis patients showed similar T1/2 (14.3 hours vs 14.0 hours), Cmax (73.6 ng/mL vs 73.3 ng/mL) and Tmax (1–2 hours according to dialysis modality vs 1.5 hours) but greater AUCinf (794 hours*ng/mL vs 558 hours*ng/mL) in contrast to patients without CKD. After 1-week administration, the mean Cmin in dialysis patients treated with henagliflozin 5 mg/day was higher than in patients without CKD treated with 10 mg/day (15.0 ng/mL vs 11.6 ng/mL). In addition, blood concentration of henagliflozin was decreased by 1.1% after a session of 4-hour HD and there were no treatment-related serious adverse events (SAEs) or drug discontinuation. These data suggest that the prolonged use of henagliflozin with a reduced dose may be safer in dialysis patients. Based on the results, we designed the current trial to explore whether the use of henagliflozin improves cardiac structural parameters and HFpEF in dialysis patients.
Objectives
The primary objective of this trial is to evaluate the effect of henagliflozin on changes in echocardiography-measured left ventricular mass index (LVMI) in patients undergoing chronic dialysis with HFpEF after 24 weeks of treatment. The secondary interests include changes in left atrial volume index (LAVI), E/e’, e’ and N-terminal pro-B-type natriuretic peptide (NT-proBNP) before and after the intervention.
This publication describes the study design and protocol of a multicentre, randomised, double-blind, placebo-controlled trial investigating the effect of henagliflozin on changes in LVH in patients undergoing chronic HD or PD complicated with HFpEF (HELD-HF study, version 1.3.1; date: 23 September 2023). Recruitment started in September 2023 and ended on 29 February 2024.
Methods and analysis
Trial design
This is a multicentre, randomised, double-blind, placebo-controlled trial. There are two parallel arms with equal allocation, including the treatment group (henagliflozin 5 mg/day in addition to standard therapy) and the control group (placebo with standard therapy). Central randomisation was employed. The enrolled participants are assigned to either group in a 1:1 ratio by a randomisation procedure based on the network stochastic system, stratified according to dialysis modality (HD and PD). The principles of double-blind randomisation are followed. The study was registered in Chinese Clinical Trial Registry (ChiCTR2300073169).
Study setting
The trial is being carried out in one primary centre (Renji Hospital, School of Medicine, Shanghai Jiao Tong University) and two subcentres (Shanghai Jiading District Central Hospital and Shanghai Punan Hospital). All investigators across the three centres have obtained Good Clinical Practice certificates.
Eligibility criteria
The target population of this trial is chronic HD or PD patients who fulfil the following criteria:
Inclusion criteria
Age 18–80 years.
Patients undergoing HD thrice a week or maintenance PD for at least 3 months, with adorable dialysis adequacy.
Clinical symptoms of HF accompanied by at least one of the following: (1) NT-proBNP >8000 pg/mL or BNP >300 pg/mL; (2) LVH approved by echocardiography within 12 months (LVMI >110 g/m2 in a male patient or >90 g/m2 in a female patient); and (3) admission for HF within 12 months before recruitment. Meanwhile, LVEF assessed by echocardiography is ≥50%.
Clinically stable hydration status.
Stable use of anti-HF agents, for example, ACE inhibitors/angiotensin receptor blocker, ARNI, β-blockers or MRA, for at least 4 weeks.
Signed the informed consent form.
Exclusion criteria
Multiple episodes of hypoglycaemia during the past 4 weeks.
Evidence of ketoacidosis within 3 months.
Type 1 diabetes mellitus.
Significant anaemia (haemoglobin <80 g/L), hypoalbuminaemia (serum albumin <30 g/L) or hepatic dysfunction (elevated alanine aminotransferase (ALT) or aspartate aminotransferase (AST) three times of the normal upper limit or greater).
Systemic or genitourinary tract infection, active malignancy, history of cardiac valve replacement or repairment, and persistent atrial fibrillation.
Allergy to SGLT2 inhibitors.
Pregnant and lactating women.
Participant of other clinical trials within 3 months.
Patients considered unsuitable for inclusion in this study by the investigators.
Interventions
The flow chart elucidating the trial is presented in figure 1. Participants of the treatment group are treated with oral henagliflozin (5 mg/tablet, one tablet a day, taken before breakfast), and the counterparts in the control group are treated with placebo tablets once a day. The administration will last for 24 weeks.

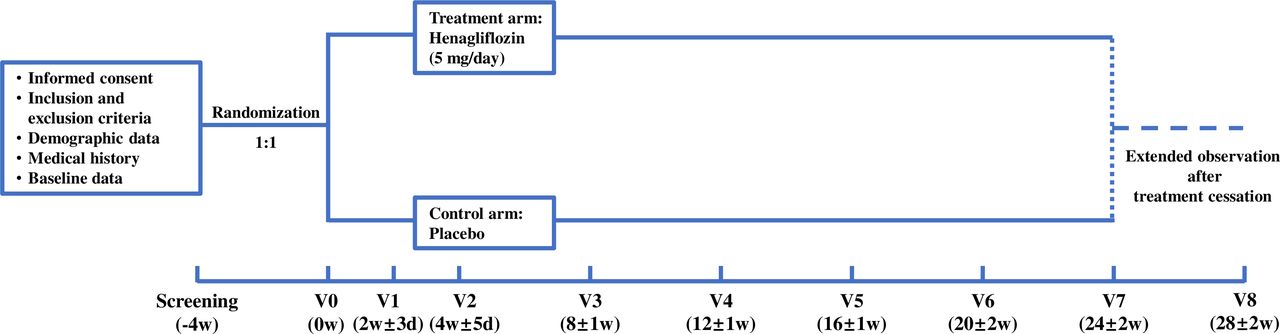{kind=link}
Participant timeline. d, days; V, vist; w, weeks.
During the follow-up, the participants will be closely monitored. If hypoglycaemia, ketoacidosis or other manifestations of intolerance occur in the participants of either group, the dose of the study drug will be reduced by half or completely withdrawn, depending on the severity and persistence, and corresponding treatments will be prescribed to correct the abnormalities. Generally, dialysis regimens and use of other anti-HF agents will remain stable while individual conditions will be taken into account. Other SGLT2 inhibitors are prohibited during the trial.
Data collection
At baseline (screening period), demographic and anthropometric data including age, sex, height and body weight are collected, as well as the medical history of the participants, such as the underlying cause of ESKD; comorbidities including hypertension, diabetes, cardiovascular disease and/or cerebrovascular disease, cirrhosis, chronic obstructive pulmonary disease, etc; dialysis vintage; dialysis prescription; urine volume; and concomitant medications.
Echocardiography and ECG are performed at baseline and 24 weeks of follow-up, accompanied by bioimpedance analysis using a body composition monitor (Fresenius Medical Care). The details of echocardiography are described in the next section. In addition, cMR is performed on subjects from the primary centre (Renji Hospital) without contraindications. Physical examination is performed at each visit, and blood samples are drawn for tests of fasting plasma glucose, pH, HCO3−, K+, Na+, Cl– and ketone body to exclude hypoglycaemia and ketoacidosis. Other laboratory parameters, including blood cell count, C reactive protein, serum albumin, ALT, AST, total bilirubin and direct bilirubin, calcium, phosphorus, haemoglobin A1c, NT-proBNP and intact parathyroid hormone, are determined at baseline and at 12 weeks and 24 weeks of follow-up, and single-pool Kt/V for HD patients or Kt/V for PD patients is measured using standard methods concurrently. Adverse events (AEs), changes in dialysis prescription and changes in concomitant medications are recorded throughout the study until 28 weeks after recruitment. For future ancillary studies, serum samples are collected at baseline, 12 weeks and 24 weeks, and are stored in −80℃ refrigerator.
Echocardiography
Transthoracic two-dimensional (2D) and Doppler echocardiography is performed by experienced echocardiographers not involved in the clinical care of the recruited patients. Interventricular septal thickness (IVS), posterior wall thickness (PWT) and left ventricular internal dimension (LVID) are measured at end-diastole and end-systole in accordance with the guidelines of the American Society of Echocardiography (ASE). Relevant images of 2D long-axis and short-axis views of end-diastolic and end-systolic ventricle with M-mode scans are recorded, and then a dedicated senior echocardiographer who is blinded to the identity of the participants and the study order (pre vs post) will further analyse each individual measurement to evaluate LVH and LV systolic and diastolic function.
LVM was calculated according to the ASE-recommended formula: LVM (g)=0.8×{1.04[(ΙVS+LVID+PWT)3 –(LVID)3]}+0.6. LVMI is defined as LVM/body surface area. LVEF was obtained using the modified biplane Simpson method from the apical two-chamber and four-chamber views.
Outcomes
Primary outcome
The primary outcome is the change in LVMI measured by echocardiography after 24 weeks of intervention.
Secondary outcomes
The secondary outcomes are the changes in LAVI, E/e’, e’ and NT-proBNP, as well as the intergroup differences, after 24 weeks of intervention.
Safety outcomes
The safety outcomes were changes in laboratory safety indicators and the incidence of AEs between the treatment and control groups from recruitment until 28 weeks thereafter.
Participant timeline
Please refer to table 1 and figure 1 for details of the visit schedule and participant timeline.
Trial visit schedule
Sample size
According to a previous randomised trial, the mean LVMI was reduced by −11.5 g/m2 with 13 weeks of administration of empagliflozin compared with −1.4 g/m2 for placebo in patients with high-risk type 2 diabetes mellitus.24 Thus, we assume that the mean difference in changes in LVMI will be 10 g/m2 between the treatment and the control group after 24 weeks of treatment, and with a common intergroup SD of 15 g/m2 and a dropout rate of 10% there should be a total of 108 participants, 54 in each arm, to have a power of 90% to yield a statistically significant result (two-tailed alpha=0.05). PASS V.15 was used to perform sample size calculation.
Recruitment
The primary centre (Renji Hospital) is a university teaching hospital that provides care to more than 500 maintenance HD patients and over 700 chronic PD patients. Both the subcentres are mature regional medical centres. There are a total of 1000 HD patients and 900 PD patients, allowing sufficient enrolment for the study.
The sponsor provides a trial subject insurance for patients participating in the study. The insurance provides coverage for study-related damage, which occurs during the study and until 4 weeks after cessation of intervention.
The recruitment started in September 2023 and ended in February 2024. In total, 122 patients were screened, among whom 112 were recruited. The baseline characteristics of the participants are summarised in table 2.
Baseline characteristics of the recruited subjects
Implementation of allocation and sequence generation
After the patient signs the informed consent, the research nephrologist performs screening and enrols eligible subjects, then sends a request to conduct randomisation. A clinical trial coordinator (CRC) uses an interactive web response system to assign an exclusive sequence number to the participant and allocates the patient to one of the study arms, stratified by dialysis modality (HD and PD). Then a dedicated research nurse distributes the appropriate trial medication (henagliflozin or placebo) corresponding to the sequence number to the participant.
Concealment mechanism
The henagliflozin and placebo tablets are indistinguishable in package, appearance and taste, preventing identification of the intervention.
Blinding and unblinding
Participants and the research personnel in the study, including nephrologists, nurses, echocardiographers and CRC, are blinded to the trial intervention. In the case of a suspected unexpected serious adverse reaction, the principal investigator (PI) is permitted to request a code break for the unblinding of the intervention allocation.
Data management
Data will be derived from electronic hospital information system. Laboratory tests are performed by the clinical laboratory in each centre. All the echocardiographers were trained according to the study requirements, ensuring that the echocardiography measurements are standardised.
The participants are closely followed up, and the contact information of the research nephrologist in charge is provided. At each visit, the participants are asked to return untaken tablets to evaluate adherence.
All data are collected by the investigators and handled in an electronic data capture system exclusively developed for the study. All changes to the raw data in the system are automatically recorded. The data set will be kept for 10 years.
Statistical methods
Data will be analysed using SPSS V.25. Analyses will follow the intention-to-treat principle, where all participants randomised are included in the group to which they were originally allocated. Data are reported as frequencies and percentages for categorical variables, mean±SD for normally distributed continuous variables and median (IQR) for non-normally distributed continuous variables. Intergroup comparison of change in LVMI from baseline to 24 weeks as the primary analysis is conducted based on a linear regression model adjusted for baseline values (analysis of covariance). For secondary analyses, the same method is used to compare LAVI, E/e’ and e’ between baseline and 24-week visits, and repeated measure analysis of variance with Bonferroni adjustment is employed to compare change in NT-proBNP from baseline to 24 weeks. Subgroup analyses of the primary and secondary outcomes are conducted to determine whether the effect of henagliflozin varies according to dialysis modality. The χ2 method is used to compare the occurrence of AEs and SAEs. In case of missing data, they are imputed using multiple imputations.
Composition of the coordinating centre and trial steering committee
The coordinating centre and the steering committee for the study consists of the PI from the primary centre (LG) and sub-PIs from the subcentres (YL and YQ). They are responsible for preparation of the protocol and any revisions, preparation of data collection and drafting of the research consent forms.
The trial management team includes the PI, sub-PIs and the senior investigators (HY, RL, YZ, SL, HP, YF and ZL). The members meet once every week to monitor and supervise the progress of the trial, review relevant information, resolve any problems that arise during the study, ensure that the protocol is adhered to and take appropriate action to safeguard participants and the quality of the trial itself.
Data monitoring
A data monitoring committee (DMC) is composed of clinicians and biostatisticians from the Clinical Center for Investigation, Renji Hospital, School of Medicine, Shanghai Jiao Tong University. The members are not involved in medical care of the study participants and are independent from the sponsor and competing interests to ensure the quality of study data. No interim analysis is planned.
Harms
Continuous monitoring and documentation of any AE will be carried out throughout the trial. All AEs reported spontaneously by the subject or observed by the investigator will be recorded and evaluated for severity and relatedness to the investigational product. SAEs are reported to the Ethics Committee of Renji Hospital without undue delay after obtaining knowledge of the events.
Auditing
Formal audits will be conducted at the request of the DMC. Representatives from the Clinical Center for Investigation, Renji Hospital, School of Medicine, Shanghai Jiao Tong University can examine trial records and personal health information to verify the accuracy of collected data. The process will be independent from investigators and the sponsor.
Patient and public involvement
None.
Ethics and dissemination
Research ethics approval
This trial has been approved by the Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University (LY2023-127-B), as well as by the ethics committees of Shanghai Jiading District Central Hospital and Shanghai Punan Hospital.
Protocol amendments
Any amendments related to the study protocol will be submitted and approved by the ethics committee of the primary centre and subcentres.
Consent
Research nephrologists had a conversation with candidates in outpatient clinic, and the interested patients were provided an information brochure with details about the trial, including the aims, methods, potential risks and benefits for the participants. The patients made decision on the consent at their discretion. Since biological specimens including blood and PD effluent are collected for future ancillary studies, there is an additional consent included in the major one.
Confidentiality
All patient information will be processed according to the National Regulation on Implementation of the Investigator Initiated Trials. Participants’ identifying information and contact information are encrypted and stored in a separate database. Identifying information is accessible exclusively by the PI of each participating centre.
Dissemination plans
Subjects are entitled to public disclosure of the results of the trial based on their participation. The results of the current research will be disclosed completely in international peer-reviewed journals. Both positive and negative results will be reported.
Discussion
Patients with ESKD are exposed to the tremendous burden of cardiovascular comorbidity. As mentioned, LVH is an established indicator of HF in this population and a main concern of treatment. According to the unpublished data of our programme, cardiovascular death contributed to 44.8% of the overall mortality in PD patients between 2018 and 2022 and HF accounted for 15.2% of the cardiovascular death. It is noteworthy that in our cohort another 40.4% of cardiovascular mortality was attributed to sudden cardiac death, which is also independently associated with LVH.25 These findings, along with the results of a previous study,3 suggest that appropriate intervention towards LVH may improve cardiac outcomes among patients with ESKD.
The mechanisms of cardiac protection by SGLT2 inhibitors probably refer to natriuresis and diuresis, which lead to attenuation of fluid and sodium retention. However, this is probably a transitory effect when the treatment begins,26 27 and in patients on dialysis whose kidney function has been significantly impaired or completely diminished this effect may be negligible. On the other hand, SGLT2 inhibition of Na+/H+ exchanger-1 on cardiomyocytes reduces intracellular sodium concentration, suppressing the Na+/Ca2+ exchanger activity and reducing cytoplasmic Ca2+ levels, subsequently inhibiting Ca2+-mediated cardiomyocyte injury.28 In addition, there is evidence of impaired myocardial energetics in HFpEF,29 which drives massive LVH,30 while SGLT2 inhibition improves myocardial energetic metabolism31; thus, it is possibly another mechanism to attenuate LVH. These give a hint to a potential role of SGLT2 inhibitors in treating LVH and consequently HFpEF among patients with ESKD.
Despite the significance of SGLT2 inhibitors in improving cardiac function and ameliorating LVH in general population, patients with advanced kidney disease have been excluded from previous studies, and the effects of SGLT2 inhibitors in chronic dialysis patients are largely unknown.
The present trial is designed to explore whether the use of henagliflozin for 24 weeks improves cardiac structural parameters and functional status of HD and PD patients with HFpEF, as well as to further validate the safety of the treatment. The dose of intervention is based on our pharmacokinetic study in dialysis patients.22 LVMI is a validated marker of HFpEF, and other echocardiographic measurements also provide important information on HFpEF, including E/e’ and LAVI, which indicate instantaneous and chronic LV filling pressures, respectively, and e’, which is a direct marker of diastolic function. Therefore, these parameters are selected as the primary and secondary endpoints in the present trial. In addition, NT-proBNP is another secondary endpoint. In long-term dialysis patients, BNP and NT-proBNP levels are strongly associated with LVH and RKF, and the best cut-off values to discriminate LVH or HF in dialysis patients have not been determined.32 We adopt either NT-proBNP >8000 pg/mL or BNP >300 pg/mL as one of the inclusion criteria according to the results of previous research33 34 as well as our clinical observation.
It is noteworthy that the designed outcomes of the trial consist of merely surrogate markers for cardiac functional status; therefore, we cannot address the question whether SGLT2 inhibitor reduces the risk of hard endpoints including mortality, new-onset HF and other cardiovascular diseases. Physical tests objectively evaluating cardiac function, for example, 6 min walk test, are also absent in the trial. However, to the best of our knowledge, there are very few ongoing randomised controlled trials investigating the effects of SGLT2 inhibitors on cardiac geometric and functional alterations among patients with ESKD, which specifically include only HD patients (NCT06249932, NCT06249945). These efforts could clarify the role of SGLT2 inhibitors in treating HF among dialysis patients and could lead to an updated evidence-based approach to managing this condition.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank the patients for their time and willingness to participate in this trial, as well as Haifeng Zhang, Jiaying Huang, Yanna He, Na Li, Aiping Gu, Xiaojun Zeng, Tingting Liu, Ping Li, Nina Fang, Hongying Tang, Ting Qiu, Yue Yu and Sijie Ren, research nurses, for their support.
References
Footnotes
Contributors LG is the guarantor of this work, and is responsible for the study design, including drafting, revising, submission and registration of the protocol. WF, JP, MJ and WZ discussed the protocol and provided important advice. YL and YQ are sub-PIs of the subcentres. JP and MJ coordinate the multidisciplinary network. WW takes charge of designing the imaging tests. JW is responsible for the design of statistical methods. HY, RL, YZ, SL, HP, YF and ZL are investigators conducting the trial. HY drafted the manuscript. All authors have read and approved the final manuscript.
Funding This trial was funded by the Program of Shanghai Academic/Technology Research Leader (22XD1431400) and Shanghai Hengrui Pharmaceuticals. The funders do not have a role in the collection, analyses and interpretation of the data or in writing the manuscript.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.