Article Text

Abstract
Introduction Women characterised by diminished ovarian reserve are considered to have poor ovarian response (POR) according to Patient-Oriented Strategies Encompassing IndividualizeD Oocyte Number (POSEIDON) criteria. Patients in this population often have a poor prognosis for treatment with assisted reproductive technology. In previous studies, oestrogen pretreatment before ovarian stimulation has been shown to have a beneficial effect. However, recent studies presented conflicting conclusions. This study aims to evaluate the effectiveness of oestrogen pretreatment in patients with expected POR (POSEIDON groups 3 and 4) undergoing gonadotrophin releasing hormone antagonist (GnRH-ant) protocol.
Methods and analysis A prospective superiority randomised parallel controlled trial will be conducted at a tertiary university-affiliated hospital. A total of 316 patients will be randomly divided into two groups at a ratio of 1:1. In the intervention group, oral oestrogen pretreatment will be administered from day 7 after ovulation until day 2 of the next menstrual cycle. Afterwards, a flexible GnRH-ant protocol will be initiated. The control group will receive no additional intervention beyond routine ovarian stimulation. The primary outcome is the number of oocytes retrieved. Secondary outcomes include the total number of retrieved metaphase II oocytes, average daily dose of gonadotropin, total gonadotropin dose and duration of ovarian stimulation, cycle cancellation rate, top quality embryos rate, blastocyst formation rate, embryo implantation rate, clinical pregnancy rate, early miscarriage rate and endometrial thickness on trigger day. All data will be analysed according to the intention-to-treat and per-protocol principles.
Ethics and dissemination The ethical approval has been confirmed by the reproductive ethics committee of the affiliated hospital of Shandong University of Traditional Chinese Medicine (SDUTCM/2022.9.20). In addition, written informed consent will be obtained from all the participants before the study. The results will be disseminated via publications.
Trial registration number ChiCTR2200064812.
- Reproductive medicine
- GYNAECOLOGY
- Clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is the first study to assess the effectiveness of oestrogen pretreatment on the number of oocytes retrieved in expected poor responders as defined by the POSEIDON criteria.
This exploratory study will provide insight and a research basis for evaluating the effects in POSEIDON groups 3 and 4.
The study is open-label and neither participants nor clinicians are blinded.
This study is conducted in a single centre to reduce bias caused by differences in clinician preferences and stimulation strategies.
In the POSEIDON groups 3 and 4, subgroup analyses may be underpowered to detect differences.
Introduction
In assisted reproductive technology (ART), poor ovarian response (POR) to controlled ovarian stimulation (COS) remains a serious dilemma. Previous studies have been limited by the inconsistent definition of poor responders, which impairs the management of poor responders and the design of clinical trials. In 2011, The European Society of Human Reproduction and Embryology (ESHRE) Bologna criteria were established, as an attempt to standardise the definition of POR and make studies comparable.1 However, the bologna criteria have been criticised, for not taking the effect of age on oocyte quality and ovarian sensitivity to exogenous gonadotropins (Gn) into consideration, which leads to subpopulations with varied baseline characteristics.2 POSEIDON established more stratified classifications of the POR population in order to establish more homogeneous clinical management for poor responders. According to the criteria, patients were stratified into two groups: the ‘unexpected’ poor responders (groups 1 and 2) and the ‘expected’ poor responders (groups 3 and 4).3 Of these, expected poor responders are defined as women aged with anti-Müllerian hormone (AMH) levels less than 1.2 ng/mL and/or antral follicle count (AFC) less than 5. Several studies have indicated that expected poor responders (POSEIDON groups 3 and 4) had fewer oocytes retrieved and poorer pregnancy outcomes than unexpected poor responders and non-POSEIDON patients.4 5
Therefore, it is crucial to determine the optimal COS protocol for those with expected poor response so that their remaining ovarian reserve can be fully used. As noted in the ESHRE 2019 guideline on ovarian stimulation for in vitro fertilisation (IVF)/intracytoplasmic sperm injection (ICSI), no differences exist in terms of safety and efficacy between the gonadotrophin releasing hormone antagonist (GnRH agonist/GnRH-a) and the GnRH-ant protocol in women with POR.6 In more specific subgroups of POR, this recommendation may be controversial. Recent studies have demonstrated that the GnRH-ant protocol produces fewer retrieved oocytes, lower cumulative pregnancy rates and lower cumulative live birth rates than the GnRH-a protocol in patients with poor responses as defined by POSEIDON.7–9 The advantage of GnRH-a protocol may be attributed to follicular synchronisation obtained after pituitary downregulation, which is paramount for expected poor responders as they usually have increased follicle stimulation hormone levels in the late luteal phase thus resulting in the large leading follicle in early recruitment and poor response of other left follicles to ovarian stimulation.10 As an adjunct to the GnRH-ant protocol, oestrogen pretreatment can also be used to achieve synchronisation through negative feedback.11–13 Additionally, oestrogen pretreatment of POR patients has been reported in several studies to improve the prognosis.14–16 Based on existing evidence and clinical practice, the POSEIDON group developed an expert opinion algorithm, in which the luteal phase oestrogen pretreatment is considered the first choice in the management of expected poor responders, and equally recommended with GnRH-a long protocol.17 While this recommendation represents the current best clinical practice, more randomised controlled trials (RCTs) are still required to provide robust evidence, given the insufficiency of existing studies.
Therefore, we intend to design an RCT to comprehensively evaluate the effectiveness of oestrogen pretreatment in GnRH-ant protocol to improve oocyte yield and pregnancy outcomes for expected poor responders. This study will contribute to existing work and guide future RCTs.
Methods and analysis
Study design
The study is a prospective, randomised, superiority, parallel, controlled trial. This RCT will be conducted at the reproductive centre of integrative medicine, the affiliated hospital of Shandong University of Traditional Chinese Medicine (SDUTCM). Patient enrolment is anticipated to begin in November 2022 and end in November 2024. The study protocol adhered to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guideline for protocols of RCTs. The study flow chart is shown in figure 1, and the study process schedule is shown in table 1.

Flow chart of the trial. GnRH, gonadotrophin-releasing hormone; ICSI, intracytoplasmic sperm injection; IVF, in vitro fertilisation.
Schedule of the study process
Eligibility criteria
Inclusion criteria
Married women aged 20–45 years diagnosed with infertility. (The requirement for marriage is due to China’s current access regulations for ART).
The POSEIDON criteria will be introduced to screen patients with expected POR (ie, groups 3 and 4), which are characterised by diminished ovarian reserve (AFC<5 or AMH<1.2 ng/ml). Patients belong to POSEIDON group 3 if they are aged < 35 years or POSEIDON group 4 if they are ≥35 years old.
Women scheduled for IVF or ICSI cycle.
Exclusion criteria
Women with any of the following exclusion criteria are not eligible to participate in the study:
Age >45 years.
Body mass index (BMI)≥35 kg/m2.
Recipients of donated oocytes.
Preimplantation genetic testing cycle.
Untreated moderate to severe endometriosis, submucosal fibroids, multiple endometrial polyps, pelvic inflammatory disease, uterine anomalies, Asherman syndrome and hydrosalpinx.
Dropout and discontinuation criteria
Those patients who withdraw from the study on their own initiative, who experience poor compliance during the study period or who are deemed unsuitable for further participation in the RCT by the investigator will be removed from the study. The reasons include, but are not limited to the following: (1) participants are pregnant during the pretreatment cycle; (2) participants experience adverse reactions or complications during the study period; (3) participants do not take estradiol valerate tablets at the prescribed dosage or duration for pretreatment and (4) participants take other medications during the study that may interfere with the study results, such as oral conceptive pills and growth hormones. Moreover, patients who have their cycles cancelled due to abnormal follicle size or hormone levels will be rescheduled for oestrogen pretreatment and IVF/ICSI.
Study population and recruitment
The participants will consist of 316 women with POR. Women who may be eligible will be recruited by the clinicians in the clinic of the reproductive centre and informed in detail about the protocol on their first visit. Once the decision to participate in the trial is made, the eligible patients will be required a secondary visit on days 2–4 in the follicular phase of the pretreatment cycle. After counselling and explanation, the participants will be required to sign the informed consent and the baseline characteristics will be evaluated. Given the high incidence of POR and the sufficient number of patients with daily visits, we will not take other additional approaches to recruitment.
Patient and public involvement
The development of this study protocol is without patient or public involvement. Patients will not be involved in the study design or study procedures. Our randomisation and treatment protocols will not be influenced by patient preferences. The final study results will be disseminated to participants on request.
Randomisation, allocation and blinding
Randomisation and allocation will begin on day 7 after the ovulation or follicular luteinisation. Eligible participants will be stratified by female age (<35 years old or ≥35 years old) and randomised within two POSEIDON groups. The participants will be randomly assigned to either the oestrogen pretreatment group or the control group in a 1:1 ratio. Randomisation will be generated by computer (R V.4.0.0, R Foundation for Statistical Computing, Vienna, Austria), and different sized block groups of 2, 4, 6 will be applied. Randomisation sequences will be completed by a statistician not involved in the rest of the study, and random numbers will be sealed into opaque envelopes in which the groups will be completed by opening the envelopes sequentially according to the order of patient recruitment.
A double-blind study design has been considered to enhance the quality of the study. However, prior studies have indicated that hormone levels and follicle size were significantly different in the oestrogen pretreatment group than in the control group before COS was initiated.13 It is almost impossible for clinicians not to notice the difference. On this basis, the study design is open-label, which means patients, researchers and clinicians are not blinded to treatment allocation. Nevertheless, the allocation is blinded to the clinician who performs the oocyte retrieval and embryo transfer. The embryologist who performs the laboratory assessment as well as the statistician who analyses the data will also be blinded.
Interventions
Oestrogen pretreatment
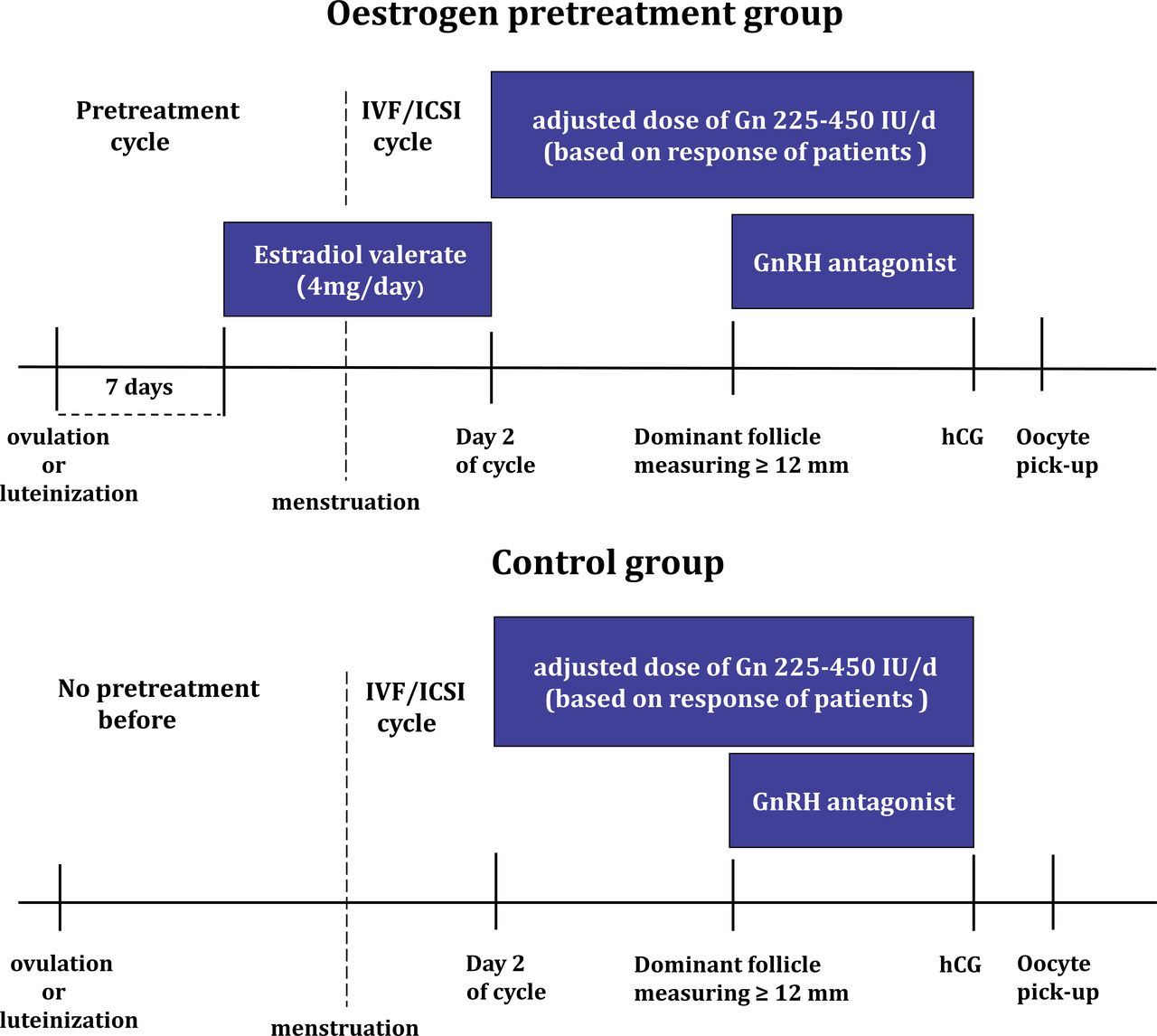
In the oestrogen pretreatment group, estradiol valerate (2 mg, Progynova, Schering AG, Berlin, Germany, two times per day) will be administered during the menstrual cycle preceding the IVF/ICSI. The ovulation date will be determined based on transvaginal ultrasound monitoring and hormone levels. The disappearance of dominant follicles observed on transvaginal ultrasound will be defined as ovulation. If there are visible light bands in the follicle, combined with a rise in serum progesterone (P4), follicular luteinisation will be confirmed. And on day 7 after ovulation or follicular luteinisation, patients will be administered oral estradiol valerate until day 2 of their next menstrual cycle.
In the control group, no pretreatment and other additional medication will be administered preceding the IVF/ICSI. The comparison between the two groups is represented in figure 2.

{kind=link}
{kind=link}
The protocols of two groups. Gn, gonadotropin; GnRH, gonadotrophin-releasing hormone; hCG, human chorionic gonadotrophin; ICSI, intracytoplasmic sperm injection; IVF, in vitro fertilisation.
Ovarian stimulation and oocyte retrieval
After spontaneous menses, participants will be assessed again for exclusion criteria on days 1–3 of the menstrual cycle. Hormonal and ultrasound assessments will also be performed to ensure the feasibility of COS. The ovarian stimulation cycle will be cancelled in case of ovarian cysts >20 mm in diameter, dominant follicles >10 mm in diameter or serum P4 levels >3 ng/mL.
Eligible participants will begin ovarian stimulation with a GnRH-ant protocol on menstrual cycle days 1–3. A flexible GnRH-ant (cetrorelix acetate, Cetrotide; Merck Serono, Darmstadt, Germany) will be initiated when the dominant follicle reached 12 mm in diameter at a daily dose of 0.25 mg and continue to the trigger, with 225–450 IU/day of recombinant follicle-stimulating hormone (75 IU, Puregon, MSD, Courbevoie, France; Gonal-F, Merck-Serono, Lyon, Italy) and recombinant luteinising hormone (75 IU, Luveris, Merck-Serono, Germany). The starting dose of Gn will be determined based on individual patient baseline characteristics, such as age and BMI. The COS process will be individualised, and the type and follow-up dose of Gn will be adjusted according to the participants’ response during ovarian stimulation. Participants will undergo serial transvaginal ultrasound follicle measurements and blood tests of serum oestradiol (E2), P4 and LH levels during the COS cycle to be dynamically measured. When more than two leading follicles reach at least 18 mm, 5000–10 000 IU of human chorionic gonadotrophin (hCG) will be administered to trigger the final follicular maturation. Approximately 35–36 hours after oocyte retrieval, transvaginal ultrasound-guided aspiration is performed.
Oocyte insemination and embryo culture
Conventional IVF or ICSI will be carried out depending on the partner’s semen quality per the standard protocols at the centre. Normal fertilisation will be assessed (a second polar body and two pronuclei) after 16–18 hours of conventional insemination or ICSI. The BD Falcon IVF medium (Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA) will be used to collect the oocytes and perform embryo culture at 6% CO2, 5% O2 and 37.0°C (C200 CO2 Incubator, Labotect Labor-Technik-Göttingen, Göttingen, Germany).
The choice of embryos for vitrification will be expected to focus on the inclusion of no less than six blastomeres with ≤20% fragmentation. Embryos that present a fragmentation rate between 20% and 50% will be transferred or vitrified when they reach the 8 cell stage on day 3. Day 5 blastocysts with a Gardner score of 3BB or higher will be considered to be good quality and suitable for transfer or vitrification.
Endometrial preparation and embryo transfer
The decision on fresh or frozen embryo transfer will be made based on the patient’s endometrial thickness, serum P4 level, embryo score and personal requirements. Embryo transfer is performed on day 3 after the oocyte pick-up day, and one or two good-quality embryos will be transferred by transvaginal ultrasound guidance. The luteal phase support (LPS) will commence on the day of oocyte retrieval.
In a frozen-thawed embryo transfer (FET) cycle, hormone replacement treatment will be used for endometrial preparation. In addition, treatment with estradiol valerate will commence on days 1–3 of the menstrual cycle at a dose of 4–6 mg per day for 10–12 days. When the endometrial thickness exceeds 8 mm, LPS treatment will be added, and FET will be performed after 3 days or 5 days depending on the stage of the surplus frozen embryo. The serum β-hCG levels will be checked 14 days after embryo transfer.
Among participants undergoing embryo transfer, LPS will continue until 10 weeks of gestation, with an intramuscular P4 injection of 40 mg per day, plus dydrogesterone tablet (Duphaston, Abbott, Hoofddorp, Netherlands) 30 mg per day or vaginal P4 (8% Crinone, Merck Serono, Switzerland) 90 mg per day. The LPS will be discontinued if the serum pregnancy test is negative or the transvaginal ultrasound reveals pregnancy failure.
Outcomes
The primary outcome is the number of oocytes retrieved per COS cycle.
In order to comprehensively evaluate the effectiveness of the oestrogen pretreatment, the following secondary outcomes will also be compared between the two groups:
The total number of retrieved metaphase II oocytes per COS cycle.
Average daily dose of gonadotropin per COS cycle.
Total gonadotropin dose and duration of ovarian stimulation per COS cycle.
Cycle cancellation rate per COS cycle.
Top quality embryos (TQE) rate per embryo. TQE is defined as embryos with more than six even blastomeres and <20% fragmentation.
Blastocyst formation rate per embryo defined as the number of blastocysts divided by the number of cleavage embryos.
Embryo implantation rate per embryo defined as the percentage of the number of the gestational sac to the total transferred embryos.
Clinical pregnancy rate per embryo transfer defined as the number of identified clinical pregnancies divided by the number of embryo transfer cycles (fresh or frozen). Clinical pregnancy is confirmed when one or more gestational sacs are detected on transvaginal ultrasound scanning.
Early miscarriage rate per patient defined as the percentage of participants with loss of a diagnosed clinical pregnancy before 12 weeks gestation to the total patients randomised.
Statistical analysis
Sample size calculation
Sample size calculations refer to a recently published study, adjusted with data from our reproductive centre.18 The study was designed to detect a difference between the two groups in terms of one additional retrieved oocyte after hCG trigger at p=0.05 and power=80%, with an SD of 3 retrieved oocytes. Assuming a 10% drop-out rate, the sample size of 316 was determined using PASS V.15.0 (NCSS), in which 158 participants were in each group.
Statistical methods
Baseline demographics and ovarian stimulation characteristics will be compared between groups using Student’s t-test, Mann-Whitney U-test or χ2 test, according to the type of variables. Intention-to-treat analysis will be used to assess the effect of oestrogen pretreatment on the total number of retrieved oocytes per COS cycle and other secondary outcomes in both groups. Normally distributed continuous variables are expressed as mean±SD. Non-normally distributed continuous variables are expressed as median and range. T-test or Mann-Whitney U test will be used to test for differences according to the distribution of the variables. Categorical variables will be expressed as frequencies and percentages, and differences will be assessed using χ2 test. A two-sided p<0.05 is considered statistically significant with a CI of 95%. Two sets of sensitivity analyses will be undertaken for the primary outcome. First, we will analyse the per-protocol population, including those patients who completed their randomised treatment. Second, the van Eiteren test adjusted for age stratum (<35 years old or ≥35 years old) will be performed. Subgroup analysis will be performed within POSEIDON group 3 and group 4 using a similar approach to the primary analysis. Additionally, missing data values will be addressed using multiple imputations and all statistical work will be performed on SPSS V.26.0 software (IBM) and R statistical package version V.4.0.0.
Data management
The baseline characteristics data, as well as information collected at time points during the pretreatment cycle and IVF/ICSI cycle, will be recorded on the day of the participant’s visit. The embryological laboratory data will be recorded on the day of embryo transfer. Three months will be required to complete the follow-up of this study. Generally, participants will visit regularly per the routine of the reproductive centre. The results of early pregnancies will be recorded via a telephone follow-up for patients not following up as scheduled. Data will be collected, entered and coded into a standard case report form (CRF) using the double-entry method. CRFs will be kept in a locked filing cabinet in the reproductive centre while the electronic CRFs will be kept in encrypted files on a password-protected computer. Only the principal investigators will have access to the files and data. All data will be deidentified by CRFs and will be stored for 10 years. Electronic data will be formatted to be removed after that period.
Data monitoring
All data will be supervised and monitored by a data monitoring committee (DMC). The DMC consists of independent physicians and statisticians from the affiliated hospital of SDUTCM. DMC will check and review the quality of data collected in the study and be responsible for interim analysis and adverse events. Throughout the study, DMC will monitor the collected data and evaluate the procedure of the study regularly. Furthermore, it will provide recommendations for continuing the clinical trial, modifying the trial protocol or terminating the trial.
Ethics and dissemination
The ethical approval has been confirmed by the reproductive ethics committee of the affiliated hospital of SDUTCM (Identifier: SDUTCM/2022.9.20). The safety profile of this study is considered high. Estradiol valerate tablet is a drug commonly used in reproductive centres and gynaecology clinics. In our centre, the drug has been optionally used for pretreatment of ovarian stimulation cycles, and no side effects have been reported as a result, and we used the same dosage (2 mg two times per day day) for our study. All of the medications and dosages used in ART cycles are routine, including COS, drug triggers and LPS. As all medication used complies with the standards, the possibility of serious adverse reactions (SARs) or suspected unexpected SARs (SUSARs) is expected to be low. Nevertheless, if a SAS or a SUSAR occurs. The treatment will stop immediately and the events will be reported to the committees within 24 hours. Then the participants will be considered drop-outs from the study.
Other than transportation, there are no additional financial costs for study participants. The trial protocol is performed following the principles of the Declaration of Helsinki and the International Council for Harmonisation Guidelines for Good Clinical Practice and applicable regulatory requirements. Written informed consent is obtained from each couple before randomisation and allocation.
The results of this study will be disseminated by publication in peer-reviewed international scientific journals targeting reproductive medicine and at chictr.gov.cn.
Discussion
Pretreatment with oestrogen before GnRH-ant cycles is considered to be one of the best treatments for poor responders.17 19 The expected poor responders take up about 65% of the POSEIDON population.20 As a result of their age, these patients are further classified into POSEIDON groups 3 and 4. Patients with diminished ovarian reserve have very few developing follicles and oocytes retrieved, which will negatively impact their chances of giving birth to a live child.21 Consequently, the primary aim of this study is to determine whether pretreatment with oestrogen in the GnRH-ant protocol can enhance oocyte retrieval and subsequent pregnancy. Up to now, clinical studies using the POSEIDON criteria are still relatively few and high-quality evidence is still lacking, especially for RCTs.22 23 To our knowledge, this study is the first RCT protocol to investigate the effect of oestrogen pretreatment following GnRH-ant protocol on reproductive outcomes in expected poor responders defined by POSEIDON criteria. To ensure comparability within each age stratum, we will perform prespecified stratified randomisations. Subgroup analyses will be conducted in order to determine the impact of the pretreatment in the two age strata. We hypothesise that the reproductive outcomes in the two strata are different, because of the age-related embryo/blastocyst aneuploidy rate, which could dramatically vary the prognosis in patients with diminished ovarian reserve at the same level.24 25 A previous study has shown that POSEIDON group 4 requires more mature oocytes to get a euploid blastocyst compared with group 3.26 Pretreatment of expected poor responders significantly increases the number of oocytes obtained. However, in subgroups, pretreatment is more effective among younger patients (POSEIDON group 3) than older patients (POSEIDON group 4).
A recent RCT conducted by Zhang et al revealed that oestrogen pretreatment in GnRH-ant protocol did not increase oocyte yield or improve other pregnancy outcomes for poor responders as defined by the Bologna criteria.18 However, oestrogen pretreatment yielded lower FSH levels and a more synchronised follicle cohort on the first day of ovarian stimulation. This study has provided the highest-quality evidence to date on the effect of oestrogen pretreatment on POR patients. However, given the heterogeneity of the Bologna criteria used in this study, the confounding bias introduced by different baseline characteristics may potentially interfere with the study outcomes. We differ from Zhang et al in several ways, including a more homogeneous and comparable study population, and differences in COS protocol. As a result of the practices at our reproductive centre and the existing evidence, we will choose individualised doses of gonadotropin and recombinant LH supplementation.17 27 28
We acknowledge that our protocol also has limitations. First, considering the expected low prognosis of our study population, we will follow an individualised ovarian stimulation strategy rather than a fixed protocol applied to all subjects to achieve optimal outcomes. The COS protocol will be flexible and adjusted according to the baseline characteristics of the patient and the ovarian response during stimulation, which may introduce potential confounding factors to the results of the study. Second, the estimation of the study sample size was on the basis of the whole population of expected poor responders, and the subgroup analysis conducted in the POSEIDON groups 3 and 4 may be underpowered to detect the difference between the pretreatment group and the control group. Third, the comparison in subgroups will not be adjusted for multiple testing to control the type I error. Accordingly, any findings obtained in the subgroup analysis may not be reproducible and should be presented as exploratory findings that must be confirmed in a further independent RCT or meta-analysis.
In conclusion, for poor responders, there is still a lack of high-quality evidence for the effectiveness of the luteal phase oestrogen pretreatment in patients undergoing GnRH-ant protocol.6 On confirmation of efficacy, we are confident that the potential benefit will be substantial for patients with poor responses. Due to its cost-effectiveness, patient-friendliness and convenience for clinicians, this protocol is widely adopted. As a result of this study, comprehensive and reliable evidence should be provided.
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank all the participants and investigators for their contributions to this study.
References
Footnotes
Contributors J-YS and ZS conceived the trial protocol. J-YS and QH are responsible for the study design and thesis writing. J-YS, QH, Z-JW, YZ, J-NL and Q-MJ contributed to protocol development and implementation, including data collection, data management, data monitoring and statistical analysis. J-YS and ZS are responsible for the critical revision of the manuscript and the enrolment of participants. All authors contributed to the article and approved the submitted version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.